
Crystal structure of SARS-CoV-2 stem–loop 5 (SL5) (PDB id: 9E9Q; Jones CP, Ferré-D'Amaré AR. 2025. Crystallographic and cryoEM analyses reveal SARS-CoV-2 SL5 is a mobile T-shaped four-way junction with deep pockets. RNA 31: 949–960). The T-shaped four-way junction of the coronavirus SL5 structural element provides a starting point for examining the structures of larger RNA motifs and their interactions with other molecules. Image highlighting the four arms of the junction. The RNA backbone is depicted by a gray ribbon. The bases within the arms of the junction are colored respectively in blue, red, yellow, and cyan. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for Structural Bioinformatics of Nucleic Acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
As the developer of DSSR, I am thrilled to see its application in cutting-edge research across multiple disciplines. Below is a list of four recent publications that highlight how DSSR has been utilized, underscoring its versatility and significance in structural bioinformatics.
In the Geng et al. (2025) Nucleic Acids Research (NAR) paper, titled 'Revealing hidden protonated conformational states in RNA dynamic ensembles', DSSR is simply cited as follows:
All bp geometries, hydrogen-bond, backbone, stacking, and sugar dihedral angles were calculated using X3DNA-DSSR [77].
In the preprint by Gordan et al. (2025), titled 'High-throughput characterization of transcription factors that modulate UV damage formation and repair at single-nucleotide resolution', DSSR is cited as follows:
Step base stacking, base pair shift, base pair slide, interbase angle, pseudorotation angle, and sugar puckering classifications of nucleobases were computed using X3DNA-DSSR (v2.5.0)75. Base stacking was defined as the overlapping polygon area in Å2 when projecting the dipyrimidine base ring atoms (excluding exocyclic atoms) into the mean base pair plane76. The sugar ring pseudorotation phase angle of each pyrimidine was also calculated using X3DNA-DSSR as described by Altona, C. & Sundaralingam, M.77 Interbase angle was defined as sqrt(propeller2+buckle2) per the X3DNA-DSSR documentation.
Figure 6: TF Binding Induces Structural Distortion Favorable to UV Dimerization is highly informative, particularly panel (a), which illustrates the ensemble of structural parameters that predispose dipyrimidines to cyclobutane pyrimidine dimers (CPD) or 6-4 pyrimidine-pyrimidones (6-4 PP) formation. DSSR is designed as an integrated software tool, offering a comprehensive suite of structural parameters not found in any other single tool I am aware of. Despite this, the innovative use of DSSR by Gordan et al. exceeds my expectations and demonstrates its versatility.
In the preprint by Kubaney et al. (2025) from the Baker group, titled 'RNA sequence design and protein-DNA specificity prediction with NA-MPNN', DSSR is cited as follows:
On the pseudoknot subset, we evaluate additional structure‐ and reactivity‐based metrics. DSSR v2.3.241 is used to extract the ground‐truth secondary structure from the native crystal structures. For each designed sequence, RibonanzaNet predicts 2A3 reactivity profiles, from which we compute predicted OpenKnot scores (see https://github.com/eternagame/OpenKnotScore)31 using the predicted reactivity together with the DSSR ground truth.
In a recent NSMB paper from the Baker group, titled 'Computational design of sequence-specific DNA-binding proteins', 3DNA is cited as follows:
RIF docking of scaffolds onto DNA targets (DBP design step 1) Structures of B-DNA for each target (Supplementary Table 2) were generated by (1) using the DNA portion of PDB 1BC8 (ref. 60), PDB 1YO5 (ref. 61), PDB 1L3L (ref. 51) or PDB 2O4A (ref. 62) or (2) using the software X3DNA63, followed by a constrained Rosetta relax of the DNA structure.
Please note that 3DNA has been replaced by DSSR. The functionality for constructing B-DNA models, previously provided by 3DNA, is now directly available in DSSR via its fiber and rebuild modules.
In the preprint by Si et al. (2025), titled 'End-to-End Single-Stranded DNA Sequence Design with All-Atom Structure Reconstruction', DSSR is cited as follows:
Since ViennaRNA and NUPACK require secondary structures as input, we used DSSR35 to extract secondary structures from the corresponding ssDNA three-dimensional structures.
The above use cases are merely a sample of how DSSR is utilized in the scientific literature. It is reasonable to state that DSSR has emerged as a de facto standard tool within the field of nucleic acid structural bioinformatics. Overall, DSSR is a mature, robust, and efficient software product that is actively developed and maintained. I am committed to making DSSR synonymous with quality and value. Its unmatched functionality, usability, and support save users significant time and effort compared to alternative solutions.
DSSR is available free of charge for academic users. Additionally, it has been integrated into other high-profile bioinformatics resources, including NAKB, PDB-redo, and N•ESPript.
References
- Geng A, Roy R, Ganser L, Li L, Al-Hashimi HM. Revealing hidden protonated conformational states in RNA dynamic ensembles. Nucleic Acids Research. 2025;53:gkaf1366. https://doi.org/10.1093/nar/gkaf1366.
- Gordan R, Wasserman H, Chi B, Bohm K, Duan M, Sahay H, et al. High-throughput characterization of transcription factors that modulate UV damage formation and repair at single-nucleotide resolution. 2025. https://doi.org/10.21203/rs.3.rs-8197218/v1.
- Kubaney A, Favor A, McHugh L, Mitra R, Pecoraro R, Dauparas J, et al. RNA sequence design and protein–DNA specificity prediction with NA-MPNN. 2025. https://doi.org/10.1101/2025.10.03.679414.
- Glasscock CJ, Pecoraro RJ, McHugh R, Doyle LA, Chen W, Boivin O, et al. Computational design of sequence-specific DNA-binding proteins. Nat Struct Mol Biol. 2025;32:2252–61. https://doi.org/10.1038/s41594-025-01669-4.
- Si Y, Xu Y, Chen L. End-to-end single-stranded DNA sequence design with all-atom structure reconstruction. 2025. https://doi.org/10.64898/2025.12.05.692525.
Recently, I came across the paper by Mitra et al. (2025) titled "RNAproDB: A Webserver and Interactive Database for Analyzing Protein-RNA Interactions." I am glad to notice that DSSR (Lu et al. 2015) has been cited extensively in this work, as follows:
As part of the processing pipeline, multiple software is run including DSSR^12^ (base-pairing geometries, protein–RNA hydrogen bonds, and RNA secondary structure), HBPLUS^17^ (hydrogen bonds involving water molecules), ... Leontis-Westhof^27^ base pair annotations (as computed by DSSR^12^) ... The structural elements (stems, loops, hairpins, junctions, etc.) are detected using DSSR^12^ and mapped to the partial projection layout (via averaging corresponding residue coordinates)... We explored the relative abundance of different standard nucleotides (A, C, G, and U) in helical vs. non-helical regions (as computed by DSSR^12^)...We quantified the propensity of base-pairing (as detected by DSSR^12^) between different RNA bases (Fig. 3D).
This is an impressive contribution on the characterization of protein-RNA interactions. Reading carefully through the paper and its supplemental PDF, I was intrigued by the following note on a water-mediated U-U base pair missed by DSSR.
Another important aspect to discuss is RNA–RNA water-mediated interactions^33,34^. ... One such example is the CUG repeat structure from PDB ID 7Y2B^35^ (Fig. S5A). The U/U mismatches in this structure are often unable to form direct hydrogen bonds (specifically, the central U/U mismatch forms no direct hydrogen bond). Therefore, DSSR^12^ does not classify it as a base pair. However, two water molecules form water-mediated hydrogen bonds between the two U bases. ...
While DSSR internally already takes consideration of water-mediated H-bonds in the detection of base pairs, it still requires: (1) at least one direct H-bond between two base atoms or a base atom to backbone, and (2) a co-planar geometry between the two bases. The water-mediated U7-U7 pair in PDB entry 7Y2B does not fulfill condition (1): the minimal distance between the two U bases is 5 Å, which is far larger than a typical H-bonding distance. Therefore, DSSR did not classify it as a base pair.
Prompted by the observation of Mitra et al. (2025), I have added a new option (--pair-water) in the DSSR v2.5.1-2025mar19 release to allow for water-mediated base pairs to be detected. Using PDB entry 7Y2B as an example, the DSSR command and related base-pairs output are shown below.
# x3dna-dssr -i=7Y2B.pdb1 --symm --pair-water
List of 13 base pairs
nt1 nt2 bp name Saenger LW DSSR
1 1:S.U1 2:S.A13 U-A WC 20-XX cWW cW-W
2 1:S.U2 2:S.A12 U-A WC 20-XX cWW cW-W
3 1:S.C3 2:S.G11 C-G WC 19-XIX cWW cW-W
4 1:S.U4 2:S.U10 U-U -- -- cWW cW-W
5 1:S.G5 2:S.C9 G-C WC 19-XIX cWW cW-W
6 1:S.C6 2:S.G8 C-G WC 19-XIX cWW cW-W
7 1:S.U7 2:S.U7 U-U Water -- cWW cW-W
8 1:S.G8 2:S.C6 G-C WC 19-XIX cWW cW-W
9 1:S.C9 2:S.G5 C-G WC 19-XIX cWW cW-W
10 1:S.U10 2:S.U4 U-U -- -- cWW cW-W
11 1:S.G11 2:S.C3 G-C WC 19-XIX cWW cW-W
12 1:S.A12 2:S.U2 A-U WC 20-XX cWW cW-W
13 1:S.A13 2:S.U1 A-U WC 20-XX cWW cW-W
Base pair #7 is water-mediated, as shown in the molecular image below. Note that .pdb1 means biological unit 1, and the option --symm reads the two symmetry-related structures in the MODEL/ENDMDL delineated ensemble as a single structure. See the DSSR User Manual for more details.

References
-
Lu, Xiang-Jun, Harmen J. Bussemaker, and Wilma K. Olson. 2015. “DSSR: An Integrated Software Tool for Dissecting the Spatial Structure of RNA.” Nucleic Acids Research, July, gkv716. https://doi.org/10.1093/nar/gkv716.
- Mitra, Raktim, Ari S. Cohen, Wei Yu Tang, Hirad Hosseini, Yongchan Hong, Helen M. Berman, and Remo Rohs. 2025. “RNAproDB: A Webserver and Interactive Database for Analyzing Protein-RNA Interactions.” Journal of Molecular Biology, February, 169012. https://doi.org/10.1016/j.jmb.2025.169012.

Cover images provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
See the 2020 paper titled "DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL" in Nucleic Acids Research and the corresponding Supplemental PDF for details. Many thanks to Drs. Wilma Olson and Cathy Lawson for their help in the preparation of the illustrations.
Details on how to reproduce the cover images are available on the 3DNA Forum.

Structure of the human minor spliceosome pre-B complex (PDB id: 8Y7E; Bai R, Yuan M, Zhang P, Luo T, Shi Y, Wan R. 2024. Structural basis of U12-type intron engagement by the fully assembled human minor spliceosome. Science 383: 1245–1252). The protein–RNA assembly reveals the mechanisms of recognition and recruitment of several small nuclear ribonucleoproteins (snRNPs) involved in the splicing of U12-type introns. The pre-mRNA is depicted by a red ribbon, and the U12 small nuclear RNA (snRNA) by a green ribbon, with bases and Watson-Crick base pairs represented as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; the proteins are shown as gold ribbons. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

Human tRNA splicing endonuclease (TSEN) complex bound to pre-tRNAArg (PDB id: 7UXA; Hayne CK, Butay KJ, Stewart ZD, Krahn JM, Perera L, Williams JG, Petrovitch RM, Deterding LJ, Matera AG, Borgnia MJ, Stanley RE. 2023. Structural basis for pre-tRNA recognition and processing by the human tRNA splicing endonuclease complex. Nat Struct Mol Biol 30: 824–833). Cryo-EM structure of the TSEN protein assembly with pre-tRNAArg provides insights into the recognition and splicing of an intron that must be removed from the pre-tRNA before translation. The pre-tRNAArg is depicted by a red ribbon, with bases and Watson-Crick base pairs represented as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; the TSEN subunits are shown as gold ribbons. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

Systemic RNA interference defective protein 1 (SID1) in complex with dsRNA (PDB id: 8XC1; Wang R, Cong Y, Qian D, Yan C, Gong D. 2024. Structural basis for double-stranded RNA recognition by SID1. Nucleic Acids Res 52: 6718–6727). The cryo-EM structure provides a major step towards understanding the mechanism of dsRNA recognition by SID1, involving extensive interactions between basic amino-acid residues and the sugar-phosphate backbone. The dsRNA chains are depicted by red, green, blue, and yellow ribbons, with bases and Watson-Crick base pairs represented as color-coded blocks and minor-groove edges colored white: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; SID1 is shown by a gold ribbon. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

Complex of arginyl-tRNA-protein transferase 1 (ATE1) with tRNAArg and a short peptide substrate (PDB id: 8UAU; Lan X, Huang W, Kim SB, Fu D, Abeywansha T, Lou J, Balamurugan U, Kwon YT, Ji CH, Taylor DJ, Zhang Y. 2024. Oligomerization and a distinct tRNA-binding loop are important regulators of human arginyl-transferase function. Nat Commun 15: 6350). The ATE1 homodimer dissociates upon binding the peptide and forms a loop that wraps around tRNAArg. The tRNAArg is depicted by a red ribbon, with bases and Watson–Crick base pairs represented as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; ATE1 is shown by a gold ribbon and the peptide by a white ribbon. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

Structure of endoribonuclease P (RNase P) in complex with pre-tRNAHis-Ser (PDB id: 8CBK; Meynier V, Hardwick SW, Catala M, Roske JJ, Oerum S, Chirgadze DY, Barraud P, Yue WW, Luisi BF, Tisné C. 2024. Structural basis for human mitochondrial tRNA maturation. Nat Commun 15: 4683). The structure reveals the first step of human mitochondrial tRNA maturation by RNase P, processing the 5′-leader of pre-tRNA. The RNA is depicted by a red ribbon, with bases and Watson-Crick base pairs represented as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; the protein assembly is shown by the gold ribbons. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

Structure of a group II intron ribonucleoprotein in the pre-ligation state (PDB id: 8T2R; Xu L, Liu T, Chung K, Pyle AM. 2023. Structural insights into intron catalysis and dynamics during splicing. Nature 624: 682–688). The pre-ligation complex of the Agathobacter rectalis group II intron reverse transcriptase/maturase with intron and 5′-exon RNAs makes it possible to construct a picture of the splicing active site. The intron is depicted by a green ribbon, with bases and Watson-Crick base pairs represented as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; the 5′-exon is shown by white spheres and the protein by a gold ribbon. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

Complex of terminal uridylyltransferase 7 (TUT7) with pre-miRNA and Lin28A (PDB id: 8OPT; Yi G, Ye M, Carrique L, El-Sagheer A, Brown T, Norbury CJ, Zhang P, Gilbert RJ. 2024. Structural basis for activity switching in polymerases determining the fate of let-7 pre-miRNAs. Nat Struct Mol Biol 31: 1426–1438). The RNA-binding pluripotency factor LIN28A invades and melts the RNA and affects the mechanism of action of the TUT7 enzyme. The RNA backbone is depicted by a red ribbon, with bases and Watson-Crick base pairs represented as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; TUT7 is represented by a gold ribbon and LIN28A by a white ribbon. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

Cryo-EM structure of the pre-B complex (PDB id: 8QP8; Zhang Z, Kumar V, Dybkov O, Will CL, Zhong J, Ludwig SE, Urlaub H, Kastner B, Stark H, Lührmann R. 2024. Structural insights into the cross-exon to cross-intron spliceosome switch. Nature 630: 1012–1019). The pre-B complex is thought to be critical in the regulation of splicing reactions. Its structure suggests how the cross-exon and cross-intron spliceosome assembly pathways converge. The U4, U5, and U6 snRNA backbones are depicted respectively by blue, green, and red ribbons, with bases and Watson-Crick base pairs shown as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; the proteins are represented by gold ribbons. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

Structure of the Hendra henipavirus (HeV) nucleoprotein (N) protein-RNA double-ring assembly (PDB id: 8C4H; Passchier TC, White JB, Maskell DP, Byrne MJ, Ranson NA, Edwards TA, Barr JN. 2024. The cryoEM structure of the Hendra henipavirus nucleoprotein reveals insights into paramyxoviral nucleocapsid architectures. Sci Rep 14: 14099). The HeV N protein adopts a bi-lobed fold, where the N- and C-terminal globular domains are bisected by an RNA binding cleft. Neighboring N proteins assemble laterally and completely encapsidate the viral genomic and antigenomic RNAs. The two RNAs are depicted by green and red ribbons. The U bases of the poly(U) model are shown as cyan blocks. Proteins are represented as semitransparent gold ribbons. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

Structure of the helicase and C-terminal domains of Dicer-related helicase-1 (DRH-1) bound to dsRNA (PDB id: 8T5S; Consalvo CD, Aderounmu AM, Donelick HM, Aruscavage PJ, Eckert DM, Shen PS, Bass BL. 2024. Caenorhabditis elegans Dicer acts with the RIG-I-like helicase DRH-1 and RDE-4 to cleave dsRNA. eLife 13: RP93979. Cryo-EM structures of Dicer-1 in complex with DRH-1, RNAi deficient-4 (RDE-4), and dsRNA provide mechanistic insights into how these three proteins cooperate in antiviral defense. The dsRNA backbone is depicted by green and red ribbons. The U-A pairs of the poly(A)·poly(U) model are shown as long rectangular cyan blocks, with minor-groove edges colored white. The ADP ligand is represented by a red block and the protein by a gold ribbon. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
Moreover, the following 30 [12(2021) + 12(2022) + 6(2023)] cover images of the RNA Journal were generated by the NAKB (nakb.org).
Cover image provided by the Nucleic Acid Database (NDB)/Nucleic Acid Knowledgebase (NAKB; nakb.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).

DSSR produces RNA secondary structures in connect table (.ct) format. According to "RNAstructure Command Line Help: File Formats" (with slight editing):
CT File Format
A CT (Connectivity Table) file contains secondary structure information for a sequence. These files are saved with a CT extension. When entering a structure to calculate the free energy, the following format must be followed.
- Start of first line: number of bases in the sequence
- End of first line: title of the structure
- Each of the following lines provides information about a given base in the sequence. Each base has its own line, with these elements in order:
- Base number: index n
- Base (A, C, G, T, U, X)
- Index n-1
- Index n+1
- Number of the base to which n is paired. No pairing is indicated by 0 (zero).
- Natural numbering. RNAstructure ignores the actual value given in natural numbering, so it is easiest to repeat n here.
Using PDB entry 1msy as an example (see Figure 1 below):
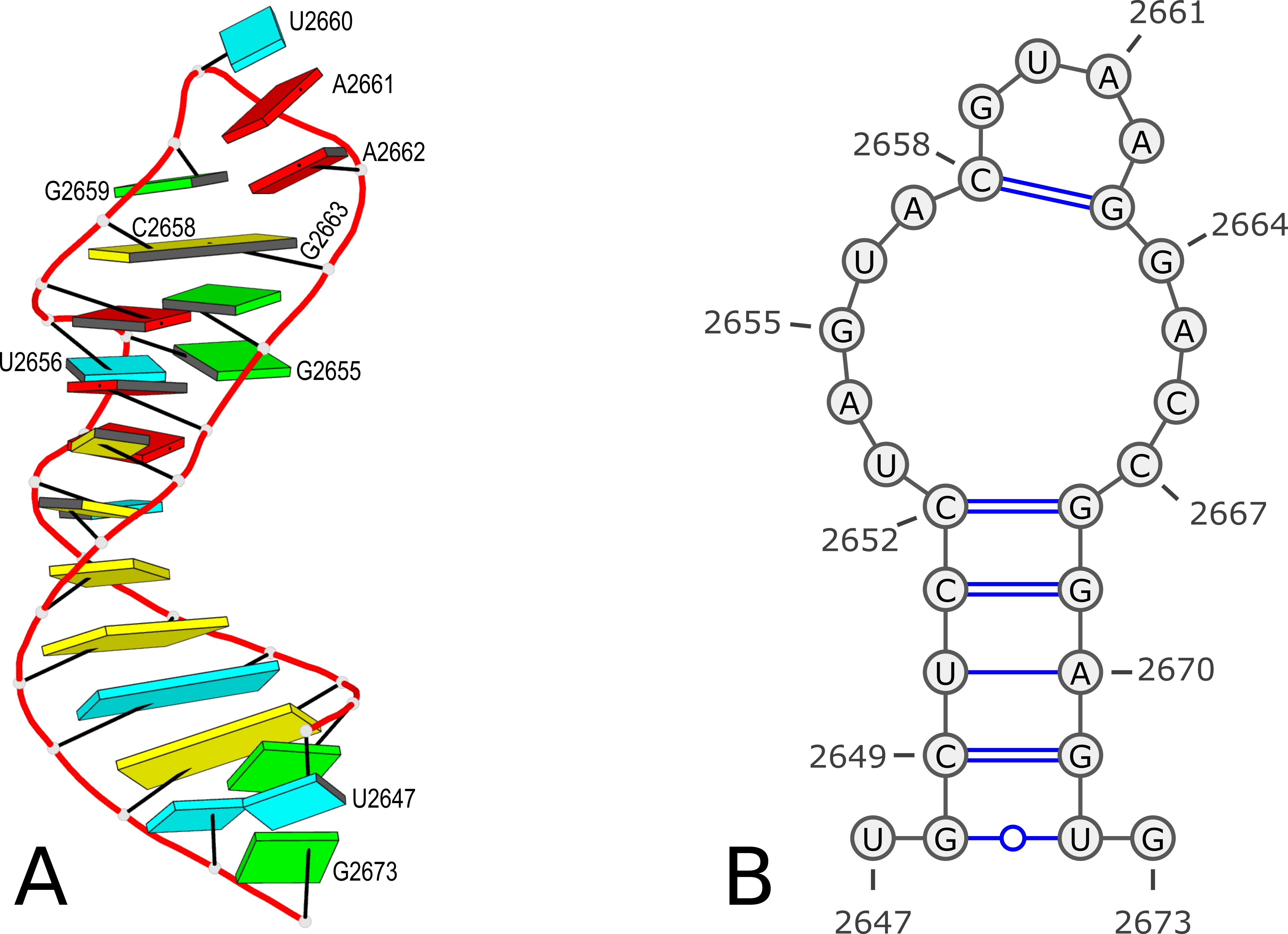
Figure 1. 3D and 2D structures of PDB entry 1msy. (A) 3D schematic auto-created via the DSSR-PyMOL integration. The labeled residues follow PDB coordinates. (B) 2D diagram rendered with VARNA using DSSR-derived 2D structural information in the .ct format. This figure was annotated using Inkscape.
With commands:
x3dna-dssr -i=1msy.pdb
cp dssr-2ndstrs.ct 1msy-dssr-default.ct
The file 1msy-dssr-default.ct has the following contents:
27 ENERGY = 0.0 [1msy] -- secondary structure derived by DSSR
1 U 0 2 0 2647
2 G 1 3 26 2648
3 C 2 4 25 2649
4 U 3 5 24 2650
5 C 4 6 23 2651
6 C 5 7 22 2652
7 U 6 8 0 2653
8 A 7 9 0 2654
9 G 8 10 0 2655
10 U 9 11 0 2656
11 A 10 12 0 2657
12 C 11 13 17 2658
13 G 12 14 0 2659
14 U 13 15 0 2660
15 A 14 16 0 2661
16 A 15 17 0 2662
17 G 16 18 12 2663
18 G 17 19 0 2664
19 A 18 20 0 2665
20 C 19 21 0 2666
21 C 20 22 0 2667
22 G 21 23 6 2668
23 G 22 24 5 2669
24 A 23 25 4 2670
25 G 24 26 3 2671
26 U 25 27 2 2672
27 G 26 0 0 2673
Here the first line contains 27 (as the number of bases) and ENERGY = 0.0 [1msy] -- secondary structure derived by DSSR (as the title). While RNAstructure ignores the actual values given in natural numbering, DSSR outputs the residue numbers of the nucleotides (e.g. U2467 and G2673) in the PDB file.
With the DSSR option --structure-title (or --str-title, actually via regex "^-?-?str(ucture)?[-_]?title"), users can set the title for the derived .ct file, as shown below:
x3dna-dssr -I=1msy.pdb --structure-title='CT file derived from DSSR'
cp dssr-2ndstrs.ct 1msy-dssr-title.ct
27 CT file derived from DSSR
1 U 0 2 0 2647
2 G 1 3 26 2648
......
26 U 25 27 2 2672
27 G 26 0 0 2673
One can also remove the title, by using an empty string "" (i.e., --str-title="") or simply --str-title (or --str-title=).
x3dna-dssr -I=1msy.pdb --structure-title=""
cp dssr-2ndstrs.ct 1msy-dssr-notitle.ct
27
1 U 0 2 0 2647
2 G 1 3 26 2648
......
With the --more option, DSSR also outputs additional info that can be used to easily identify a nucleotide and its pairing partner.
x3dna-dssr -I=1msy.pdb --more --structure-title="1msy with extra info"
cp dssr-2ndstrs.ct 1msy-dssr-extra.ct
27 1msy with extra info
1 U 0 2 0 2647 # name=A.U2647
2 G 1 3 26 2648 # name=A.G2648, pairedNt=A.U2672
3 C 2 4 25 2649 # name=A.C2649, pairedNt=A.G2671
......
Note that unlike for the .bpseq format with extra info which cannot be fed directly into VARNA, the extra info for the .ct format causes no troubles for VARNA to visualize the 2d structure.
The --structure-title option is another small feature implemented in DSSR. It is currently not documented in the DSSR User Manual since this feature is unlikely of general interest.
DSSR commands used, and the output .ct files:
x3dna-dssr -i=1msy.pdb
cp dssr-2ndstrs.ct 1msy-dssr-default.ct
x3dna-dssr -I=1msy.pdb --structure-title='CT file derived from DSSR'
cp dssr-2ndstrs.ct 1msy-dssr-title.ct
x3dna-dssr -I=1msy.pdb --structure-title=""
cp dssr-2ndstrs.ct 1msy-dssr-notitle.ct
x3dna-dssr -I=1msy.pdb --more --structure-title="1msy with extra info"
cp dssr-2ndstrs.ct 1msy-dssr-extra.ct
By default, DSSR produces RNA secondary structures in three commonly used file formats––ViennaRNA package dbn, Mfold connect table (.ct), and CRW bpseq––that can be fed directly into visualization tools such as VARNA. In this blog post, I want to dig deeper into the bpseq format, and show the variations available from DSSR.
According to "RNA STRAND v2.0 - The RNA secondary STRucture and statistical ANalysis Database" (with slight editing):
BPSEQ format:The file name should end with the suffix ".bpseq", as in "mystr.bpseq". The bpseq format is a simple text format in which there is one line per base in the molecule, listing the position of the base (leftmost position is 1), the base name (A,C,G,U, or other alphabetical characters), and the position number of the base to which it is paired, with a 0 denoting that the base is unpaired. For more information, see the Comparative RNA Web Site. An example is as follows:
1 G 8
2 G 7
3 C 0
4 A 0
5 U 0
6 U 0
7 C 2
8 C 1
For complexes with more than one molecule, the molecules are listed in sequence, with the base pairs numbers of each successive molecule following in order from the previous molecule.
The bases in bpseq format are identified by position numbers starting from 1 for the leftmost position. That is the convention DSSR follows by default in its .bpseq output. For example, for the PDB entry 1msy, which contains 27 nucleotides, the command x3dna-dssr -i=1msy.pdb will generate a file named dssr-2ndstrs.bpseq with the following contents (abbreviated):
1 U 0
2 G 26
3 C 25
......
25 G 3
26 U 2
27 G 0
However, according to PDB atomic coordinates, the nucleotides are numbered from U2647 (#1) to G2673 (#27) as shown in the Figure 1 below:
Figure 1. 3D and 2D structures of PDB entry 1msy. (A) 3D schematic auto-created via the DSSR-PyMOL integration. The labeled residues follow PDB coordinates. (B) 2D diagram rendered with VARNA using DSSR-derived 2D structural information in the .ct format. This figure was annotated using Inkscape.
It makes sense that the labelling of bases in the 2D bpseq format follows those from the 3D atomic coordinates in the PDB. Thus instead of starting from position 1 as shown above, the bpseq file would start with 2647. That's exactly what the DSSR --bpseq option is created for. Thus, with the command x3dna-dssr -i=1msy.pdb --bpseq, the output file dssr-2ndstrs.bpseq now has the following contents (abbreviated):
2647 U 0
2648 G 2672
2649 C 2671
......
2671 G 2649
2672 U 2648
2673 G 0
This .bpseq file can be read by VARNA (tested with VARNAv3-93.jar) to generate a 2D image as shown in Figure 1(B) above.
Moreover, with the command x3dna-dssr -i=1msy.pdb --bpseq=extra, the output file dssr-2ndstrs.bpseq now contains additional info to easily identify a nucleotide and its pairing partner:
2647 U 0 # name=A.U2647
2648 G 2672 # name=A.G2648, pairedNt=A.U2672
2649 C 2671 # name=A.C2649, pairedNt=A.G2671
......
2671 G 2649 # name=A.G2671, pairedNt=A.C2649
2672 U 2648 # name=A.U2672, pairedNt=A.G2648
2673 G 0 # name=A.G2673
It should be noted that this .bpseq output file is no longer compliant to the standard, and can not be fed into VARNA for visualization.
The --bpseq option has been added upon users' request. The --bpseq=extra variation was implemented recently to ensure that the --bpseq option by itself produce a valid .bpseq file without extra info (e.g., enabled with the global --more option). Now the extra info for .bpseq output is enabled only by setting --bpseq=extra explicitly.
This --bpseq option and its evolution is a good example of how DSSR responds to community requests. I'm here to listen and I'm always willing to improve DSSR that better fit users' needs. If you make use of DSSR in your pipeline and need some adaptions, please do not hesitate to contact me. I may consider adding a new option or revising the code otherwise that would streamline the integration of DSSR into your project.
DSSR commands used, and the output .bpseq files:
x3dna-dssr -i=1msy.pdb
cp dssr-2ndstrs.bpseq 1msy-dssr-default.bpseq
x3dna-dssr -i=1msy.pdb --bpseq
cp dssr-2ndstrs.bpseq 1msy-dssr-bpseq.bpseq
x3dna-dssr -i=1msy.pdb --bpseq=extra
cp dssr-2ndstrs.bpseq 1msy-dssr-bpseq-extra.bpseq
By following citations to 3DNA/DSSR, I recently came across the paper "RNAtango: Analysing and comparing RNA 3D structures via torsional angles" in PLOS Computational Biology by Mackowiak M, Adamczyk B, Szachniuk M, and Zok T. This work provides a nice summary of definitions of torsion and pseudo-torsion angles in RNA structure, and an angular metrics (MCQ, Mean of Circular Quantities) to score structure similarity. The RNAtango web application allows user to explore the distribution of torsion angles in a single structure/fragment (Single model), compare RNA models with a native structure (Models vs Target), or perform a comparative analysis in a set of models (Model vs Model).
In the Introduction section, 3DNA/DSSR are mentioned along with other related tools, as below:
Several bioinformatics tools have been designed for analyzing torsion and pseudotorsion angles, each with its own strengths and limitations. 3DNA, an open-source toolkit, provides comprehensive functionality, including torsion and pseudotorsion angle calculations [27], but lacks support for the current standard PDBx/mmCIF file format. DSSR, the successor to 3DNA, overcomes this limitation by supporting both PDB and PDBx/mmCIF files. However, it is a closed-source, commercial application that requires licensing, even for research purposes [28]. Curves+, another tool used for torsion angle analysis, is currently inaccessible due to the unavailability of its webpage and source code hosting [29]. Barnaba, a Python library and toolset for analyzing single structures or trajectories, supports torsion angle calculations but, like 3DNA, does not support the PDBx/mmCIF format [30]. For users seeking a more user friendly option, AMIGOS III offers a PyMOL plugin that calculates pseudotorsion angles and presents them in Ramachandran-like plots [17].
Every bioinformatic software has been developed for a specific purpose, and no two such tools can be identical. It is a good thing that the community has a choice for RNA backbone analysis. Indeed, 3DNA has been superseded by DSSR, which is licensed by Columbia Technology Ventures (CTV) to ensure its continuous development and availability. However, DSSR remain competitive due to its unmatched functionality, usability, and support: it saves users a substantial amount of time and effort when compared to other options.
From the very beginning, it has been my dream to make DSSR stand out for its quality and value, and be widely accessible. The CTV DSSR distribution by no means follow typical commercial license for a software product: specifically, it does not include a license key to limit DSSR's usage to a specific machine and operating system, and there is no expire date for the software either. Moreover, the Basic Academic license was free of charge when DSSR was initially licensed by the CTV in August 2020, and remained so until around end of 2021 when the web-based "Express Licenses" functionality no longer worked. Manually handling the large number of requests for free academic licenses was not sustainable, and that was when the DSSR Basic Academic free license was removed. Upon user requests, we late on re-introduced DSSR Basic Academic license, but with a one-time fee of $200 to cover the running cost. That may be reason for the remark in the RNAtango paper that DSSR "requires licensing, even for research purposes".
With the recent NIH R24 funding support on "X3DNA-DSSR: a resource for structural bioinformatics of nucleic acids", we are providing DSSR Basic free of charge to the academic community. Academic Users may submit a license request for DSSR Basic or DSSR Pro by clicking "Express Licensing". Checking the list of licensees, I am thrilled to see the many new DSSR users from leading institutions around the world, including Stockholm University, Ghent University, Universitaet Heidelberg, University of Palermo, CSSB-Hamburg, Nicolaus Copernicus University, NIH, Harvard, ... Clearly, DSSR fills a niche, and the demands for it remain strong!
Back to torsion angles, it is safe to say that DSSR has unique features not available or easily accessible elsewhere. Here are some use cases using tRNA PDB entry 1ehz as an example:
x3dna-dssr -i=1ehz.cif # generate dssr-torsions.txt among other output files
x3dna-dssr -i=1ehz.cif --torsion-file -o=1ehz-torsions.txt # just the torsion file 1ehz-torsions.txt
x3dna-dssr -i=1ehz.cif --json | jq .nts[54] > 1ehz-PSU55.txt # DSSR-derived features for nucleotide PSU55
Users can easily run the DSSR commands listed above and get the results in human-readable text and machine-friendly JSON formats. For verification, the contents of 1ehz-torsions.txt and 1ehz-PSU55.txt are available by clicking the links.
It is worth noting that DSSR has the --nmr option for the analysis of an ensemble of NMR structures, in .pdb or .cif format, as deposited in the PDB. The combination of --nmr and --json renders DSSR easily accessible to the molecular dynamics (MD) community.
In principle, calculating torsion angles is a straightforward process. In reality, factors such as modified nucleotides (especially pseudouridine), missing atoms, NMR ensembles or MD trajectories, PDB vs mmCIF formats, etc. make the implementation complicated. Without paying great attention to details, it is easy to make subtle mistakes. For example, with RNAtango the chi (χ) torsion angle for A.PSU55 of 1ehz is listed as -152.42°, which is wrong. The correct value should be -147.0° as reported by DSSR (see below and the link 1ehz-PSU55.txt above).
DSSR provides a comprehensive list of backbone parameters (as listed below for 1ehz). The program is efficient and robust, and has been battle tested. I am always quick to fix any bugs once verified, and am willing to add new features once thoroughly studied. In short, DSSR has been designed to be a reliable tool that the community can trust and build upon.
DSSR-derived backbone features for tRNA 1ehz:
Output of DNA/RNA backbone conformational parameters
DSSR v2.4.5-2024sep24 by xiangjun@x3dna.org
******************************************************************************************
Main chain conformational parameters:
alpha: O3'(i-1)-P-O5'-C5'
beta: P-O5'-C5'-C4'
gamma: O5'-C5'-C4'-C3'
delta: C5'-C4'-C3'-O3'
epsilon: C4'-C3'-O3'-P(i+1)
zeta: C3'-O3'-P(i+1)-O5'(i+1)
e-z: epsilon-zeta (BI/BII backbone classification)
chi for pyrimidines(Y): O4'-C1'-N1-C2; purines(R): O4'-C1'-N9-C4
Range [170, -50(310)] is assigned to anti, and [50, 90] to syn
phase-angle: the phase angle of pseudorotation and puckering
sugar-type: ~C2'-endo for C2'-endo like conformation, or
~C3'-endo for C3'-endo like conformation
Note the ONE column offset (for easy visual distinction)
ssZp: single-stranded Zp, defined as the z-coordinate of the 3' phosphorus atom
(P) expressed in the standard reference frame of the 5' base; the value is
POSITIVE when P lies on the +z-axis side (base in anti conformation);
NEGATIVE if P is on the -z-axis side (base in syn conformation)
Dp: perpendicular distance of the 3' P atom to the glycosidic bond
[Ref: Chen et al. (2010): "MolProbity: all-atom structure
validation for macromolecular crystallography."
Acta Crystallogr D Biol Crystallogr, 66(1):12-21]
splay: angle between the bridging P to the two base-origins of a dinucleotide.
nt alpha beta gamma delta epsilon zeta e-z chi phase-angle sugar-type ssZp Dp splay
1 G A.G1 --- -128.1 67.8 82.9 -155.6 -68.6 -87(BI) -167.8(anti) 16.1(C3'-endo) ~C3'-endo 4.59 4.57 24.92
2 C A.C2 -67.4 -178.4 53.8 83.4 -145.1 -76.8 -68(BI) -163.8(anti) 16.1(C3'-endo) ~C3'-endo 4.52 4.63 21.15
3 G A.G3 -74.5 169.7 59.5 80.7 -148.3 -80.0 -68(BI) -161.9(anti) 14.6(C3'-endo) ~C3'-endo 4.75 4.69 22.28
4 G A.G4 -64.4 162.2 60.7 82.2 -157.4 -68.7 -89(BI) -168.7(anti) 20.8(C3'-endo) ~C3'-endo 4.68 4.57 25.22
5 A A.A5 -74.7 -176.5 53.4 84.9 -137.5 -81.7 -56(BI) -162.9(anti) 4.8(C3'-endo) ~C3'-endo 4.49 4.76 22.04
6 U A.U6 -48.8 157.6 55.3 81.3 -151.0 -77.0 -74(BI) -160.0(anti) 18.2(C3'-endo) ~C3'-endo 4.31 4.51 22.89
7 U A.U7 -59.5 -178.7 62.5 137.3 -105.9 -52.0 -54(--) -133.1(anti) 156.1(C2'-endo) ~C2'-endo 1.55 1.41 126.99
8 U A.U8 -83.8 -145.6 55.4 78.6 -142.8 -118.6 -24(--) -161.5(anti) 10.5(C3'-endo) ~C3'-endo 4.60 4.76 62.37
9 A A.A9 -69.7 -141.7 52.3 147.8 -106.2 -77.3 -29(--) -70.5(anti) 149.8(C2'-endo) ~C2'-endo 1.00 1.14 57.38
10 g A.2MG10 177.8 147.2 60.1 89.3 -126.2 -88.7 -37(--) 169.6(anti) 6.6(C3'-endo) ~C3'-endo 4.68 4.63 23.87
11 C A.C11 -56.1 167.9 48.2 87.2 -150.5 -69.9 -81(BI) -160.9(anti) 16.8(C3'-endo) ~C3'-endo 4.28 4.46 21.20
12 U A.U12 -67.8 172.9 51.8 80.7 -158.5 -65.2 -93(BI) -158.3(anti) 25.2(C3'-endo) ~C3'-endo 4.29 4.45 21.01
13 C A.C13 166.6 -169.9 178.6 82.5 -153.1 -97.4 -56(BI) -168.3(anti) 23.7(C3'-endo) ~C3'-endo 4.28 4.36 31.59
14 A A.A14 83.4 -158.3 -114.6 92.0 -125.5 -57.3 -68(--) -170.7(anti) 358.9(C2'-exo) ~C3'-endo 4.67 4.74 38.01
15 G A.G15 -55.1 162.5 51.9 79.8 -136.3 -143.9 8(--) -164.5(anti) 16.0(C3'-endo) ~C3'-endo 4.72 4.74 26.17
16 u A.H2U16 -6.1 91.2 76.8 96.8 -61.8 -131.2 69(--) -85.8(anti) 18.8(C3'-endo) ~C3'-endo -0.71 3.38 145.77
17 u A.H2U17 27.8 107.7 174.1 94.8 178.0 76.2 102(--) -142.5(anti) 341.4(C2'-exo) ~C3'-endo -0.90 4.20 105.55
18 G A.G18 45.4 -159.4 59.0 150.6 -95.2 -179.1 84(BII) -99.5(anti) 154.3(C2'-endo) ~C2'-endo 1.60 1.09 51.64
19 G A.G19 -71.4 -178.9 53.8 153.8 -91.6 -83.7 -8(--) -80.3(anti) 167.6(C2'-endo) ~C2'-endo -1.14 0.48 130.30
20 G A.G20 -81.3 -150.7 47.8 89.9 -122.3 -54.1 -68(--) 177.8(anti) 8.7(C3'-endo) ~C3'-endo 4.90 4.76 57.04
21 A A.A21 -75.6 148.6 -176.6 78.2 -168.9 -75.6 -93(BI) -160.2(anti) 13.0(C3'-endo) ~C3'-endo 4.00 4.26 40.66
22 G A.G22 158.8 153.5 179.3 82.0 -145.0 -80.4 -65(BI) -175.5(anti) 353.8(C2'-exo) ~C3'-endo 4.60 4.73 25.62
23 A A.A23 -53.3 174.8 52.5 82.3 -155.3 -66.4 -89(BI) -158.0(anti) 12.6(C3'-endo) ~C3'-endo 4.18 4.61 22.96
24 G A.G24 -68.8 178.2 46.8 83.6 -144.3 -72.8 -71(BI) -160.7(anti) 13.4(C3'-endo) ~C3'-endo 4.63 4.74 20.51
25 C A.C25 -65.1 168.9 53.9 83.3 -145.1 -68.4 -77(BI) -160.3(anti) 17.4(C3'-endo) ~C3'-endo 4.56 4.70 30.70
26 g A.M2G26 -53.8 170.8 47.7 86.0 -136.3 -76.9 -59(BI) -163.4(anti) 9.3(C3'-endo) ~C3'-endo 4.57 4.67 27.36
27 C A.C27 -53.0 166.9 43.6 83.4 -148.5 -73.4 -75(BI) -168.2(anti) 18.3(C3'-endo) ~C3'-endo 4.53 4.62 23.07
28 C A.C28 -72.4 178.3 49.3 80.1 -152.1 -67.0 -85(BI) -160.6(anti) 9.2(C3'-endo) ~C3'-endo 4.55 4.73 21.61
29 A A.A29 -66.6 174.0 55.6 81.4 -155.5 -78.3 -77(BI) -165.9(anti) 13.7(C3'-endo) ~C3'-endo 4.73 4.65 26.96
30 G A.G30 -54.0 165.9 56.9 83.6 -144.7 -62.3 -82(BI) -171.7(anti) 14.5(C3'-endo) ~C3'-endo 4.67 4.65 25.72
31 A A.A31 -69.9 177.8 52.3 83.7 -137.0 -75.5 -61(BI) -156.7(anti) 14.6(C3'-endo) ~C3'-endo 4.24 4.72 21.52
32 c A.OMC32 -52.7 161.4 49.3 80.1 -145.9 -71.2 -75(BI) -149.9(anti) 20.4(C3'-endo) ~C3'-endo 4.16 4.63 25.94
33 U A.U33 -67.7 -177.0 47.0 82.1 -148.0 -53.7 -94(BI) -148.2(anti) 13.3(C3'-endo) ~C3'-endo 4.19 4.64 75.47
34 g A.OMG34 171.1 148.1 52.5 83.4 -132.5 -71.8 -61(BI) -171.2(anti) 12.2(C3'-endo) ~C3'-endo 4.15 4.58 22.09
35 A A.A35 -47.7 163.7 40.2 80.9 -143.7 -59.5 -84(BI) -154.4(anti) 21.9(C3'-endo) ~C3'-endo 4.20 4.54 20.57
36 A A.A36 -52.4 165.7 51.3 72.2 -160.4 -85.2 -75(BI) -158.4(anti) 45.8(C4'-exo) ~C3'-endo 4.49 4.31 24.48
37 g A.YYG37 -57.5 163.0 47.8 81.1 -148.1 -67.0 -81(BI) -168.8(anti) 15.4(C3'-endo) ~C3'-endo 4.63 4.65 32.08
38 A A.A38 -61.8 -180.0 46.9 82.5 -136.8 -76.4 -60(BI) -169.4(anti) 2.4(C3'-endo) ~C3'-endo 4.63 4.78 23.75
39 P A.PSU39 -47.7 160.4 53.3 79.3 -140.1 -68.6 -72(BI) -165.6(anti) 15.8(C3'-endo) ~C3'-endo 4.55 4.68 26.68
40 c A.5MC40 -67.4 172.0 56.2 83.2 -154.2 -74.9 -79(BI) -162.6(anti) 17.3(C3'-endo) ~C3'-endo 4.52 4.60 27.71
41 U A.U41 -68.2 -179.4 52.4 78.9 -137.3 -84.7 -53(BI) -169.0(anti) 13.4(C3'-endo) ~C3'-endo 4.54 4.75 24.14
42 G A.G42 -47.9 158.7 55.6 79.8 -160.3 -70.3 -90(BI) -169.0(anti) 20.9(C3'-endo) ~C3'-endo 4.43 4.51 23.54
43 G A.G43 -67.0 -178.3 55.6 81.6 -154.9 -76.4 -78(BI) -160.2(anti) 12.6(C3'-endo) ~C3'-endo 4.24 4.61 20.95
44 A A.A44 -59.7 162.1 60.0 85.3 -142.8 -57.2 -86(BI) -159.4(anti) 16.9(C3'-endo) ~C3'-endo 4.25 4.61 31.07
45 G A.G45 -71.9 -176.9 51.0 87.6 -135.1 -78.7 -56(BI) -149.3(anti) 15.4(C3'-endo) ~C3'-endo 4.01 4.58 40.27
46 g A.7MG46 -56.8 -146.5 48.4 141.6 -102.7 -137.9 35(--) -65.8(anti) 154.5(C2'-endo) ~C2'-endo 0.21 0.96 139.04
47 U A.U47 62.4 -164.0 44.4 146.1 -93.7 -78.0 -16(--) -112.0(anti) 164.9(C2'-endo) ~C2'-endo 0.26 0.39 157.37
48 C A.C48 -73.5 -174.3 161.5 145.6 -143.5 75.6 141(--) -140.1(anti) 158.2(C2'-endo) ~C2'-endo 1.92 1.80 147.54
49 c A.5MC49 50.7 168.5 42.2 84.3 -145.0 -82.1 -63(BI) -173.6(anti) 10.1(C3'-endo) ~C3'-endo 4.77 4.75 25.83
50 U A.U50 -51.7 177.2 42.1 80.4 -150.6 -67.8 -83(BI) -165.3(anti) 5.6(C3'-endo) ~C3'-endo 4.38 4.75 23.15
51 G A.G51 -63.9 176.8 52.8 79.4 -150.4 -71.3 -79(BI) -156.6(anti) 11.5(C3'-endo) ~C3'-endo 4.44 4.67 21.28
52 U A.U52 -64.7 173.6 48.5 80.3 -156.5 -69.4 -87(BI) -164.0(anti) 14.1(C3'-endo) ~C3'-endo 4.64 4.74 25.47
53 G A.G53 -56.9 171.5 56.2 83.9 -159.4 -64.9 -95(BI) -169.2(anti) 19.8(C3'-endo) ~C3'-endo 4.59 4.57 24.53
54 t A.5MU54 -79.7 -172.8 57.7 77.6 -128.6 -70.7 -58(BI) -161.5(anti) 20.6(C3'-endo) ~C3'-endo 4.56 4.80 30.73
55 P A.PSU55 -49.7 168.8 44.1 76.6 -140.8 -69.9 -71(BI) -147.0(anti) 10.1(C3'-endo) ~C3'-endo 4.15 4.74 71.28
56 C A.C56 166.4 171.8 53.3 83.4 -132.7 -70.6 -62(BI) -161.5(anti) 12.6(C3'-endo) ~C3'-endo 4.37 4.76 28.07
57 G A.G57 -65.7 167.1 57.5 81.7 -145.2 -67.6 -78(BI) -159.3(anti) 12.8(C3'-endo) ~C3'-endo 4.36 4.65 42.47
58 a A.1MA58 -60.8 -146.1 71.8 156.7 -78.3 -169.3 91(BII) -86.3(anti) 161.1(C2'-endo) ~C2'-endo 0.48 0.68 73.92
59 U A.U59 72.6 -158.8 63.7 84.6 -148.8 -53.7 -95(BI) -165.6(anti) 25.8(C3'-endo) ~C3'-endo 4.67 4.42 27.88
60 C A.C60 -72.2 179.5 66.0 148.3 -97.1 -66.4 -31(--) -117.8(anti) 154.8(C2'-endo) ~C2'-endo 0.99 0.86 90.64
61 C A.C61 -84.3 179.8 38.2 83.0 -152.3 -74.5 -78(BI) -166.7(anti) 14.8(C3'-endo) ~C3'-endo 4.45 4.52 25.80
62 A A.A62 -60.1 179.6 46.9 80.5 -145.6 -74.1 -71(BI) -158.7(anti) 9.9(C3'-endo) ~C3'-endo 4.18 4.66 19.23
63 C A.C63 -62.0 167.3 50.9 80.7 -152.3 -70.7 -82(BI) -152.6(anti) 10.7(C3'-endo) ~C3'-endo 4.32 4.62 23.62
64 A A.A64 -66.9 180.0 44.1 75.8 -147.5 -76.5 -71(BI) -161.8(anti) 12.9(C3'-endo) ~C3'-endo 4.68 4.86 25.64
65 G A.G65 -44.0 164.2 49.9 79.8 -152.0 -73.3 -79(BI) -172.8(anti) 16.5(C3'-endo) ~C3'-endo 4.92 4.76 25.20
66 A A.A66 -57.9 178.5 52.0 81.7 -151.0 -73.5 -77(BI) -164.9(anti) 22.5(C3'-endo) ~C3'-endo 4.56 4.60 22.73
67 A A.A67 -62.0 164.1 54.2 83.2 -152.2 -78.3 -74(BI) -162.8(anti) 15.0(C3'-endo) ~C3'-endo 4.71 4.67 23.30
68 U A.U68 -59.8 175.3 47.3 82.2 -152.9 -65.4 -88(BI) -160.1(anti) 11.2(C3'-endo) ~C3'-endo 4.30 4.60 24.35
69 U A.U69 -63.8 168.1 55.1 79.1 -155.4 -85.6 -70(BI) -161.4(anti) 14.7(C3'-endo) ~C3'-endo 4.55 4.61 19.23
70 C A.C70 -61.7 164.6 53.1 79.0 -158.5 -64.5 -94(BI) -152.0(anti) 15.0(C3'-endo) ~C3'-endo 4.20 4.56 20.96
71 G A.G71 -78.4 173.6 60.3 80.3 -149.6 -68.4 -81(BI) -162.8(anti) 13.5(C3'-endo) ~C3'-endo 4.50 4.71 22.80
72 C A.C72 -73.2 176.2 62.1 83.0 -152.3 -67.9 -84(BI) -161.6(anti) 19.5(C3'-endo) ~C3'-endo 4.56 4.63 26.14
73 A A.A73 -63.3 177.7 50.4 81.6 -148.2 -66.1 -82(BI) -167.4(anti) 15.0(C3'-endo) ~C3'-endo 4.65 4.71 26.33
74 C A.C74 -66.9 -174.9 50.7 85.9 -145.0 -58.8 -86(BI) -153.1(anti) 11.8(C3'-endo) ~C3'-endo 4.22 4.61 33.45
75 C A.C75 -52.3 175.7 42.3 85.6 -131.9 163.9 64(BII) -151.7(anti) 15.1(C3'-endo) ~C3'-endo 3.96 4.60 159.78
76 A A.A76 -71.0 130.2 164.6 160.9 --- --- --- 138.5(anti) 176.1(C2'-endo) ~C2'-endo --- --- ---
******************************************************************************************
Virtual eta/theta torsion angles:
eta: C4'(i-1)-P(i)-C4'(i)-P(i+1)
theta: P(i)-C4'(i)-P(i+1)-C4'(i+1)
[Ref: Olson (1980): "Configurational statistics of polynucleotide chains.
An updated virtual bond model to treat effects of base stacking."
Macromolecules, 13(3):721-728]
eta': C1'(i-1)-P(i)-C1'(i)-P(i+1)
theta': P(i)-C1'(i)-P(i+1)-C1'(i+1)
[Ref: Keating et al. (2011): "A new way to see RNA." Quarterly Reviews
of Biophysics, 44(4):433-466]
eta": base(i-1)-P(i)-base(i)-P(i+1)
theta": P(i)-base(i)-P(i+1)-base(i+1)
nt eta theta eta' theta' eta" theta"
1 G A.G1 --- -139.3 --- -136.5 --- -110.8
2 C A.C2 171.9 -144.6 -175.5 -144.1 -136.1 -118.1
3 G A.G3 160.2 -151.4 173.9 -153.9 -145.0 -143.7
4 G A.G4 164.3 -144.6 177.7 -144.1 -154.8 -98.7
5 A A.A5 166.9 -138.1 -178.3 -135.8 -116.3 -111.6
6 U A.U6 172.1 -149.7 -170.8 -143.9 -130.1 -126.5
7 U A.U7 -158.0 -42.7 -138.7 -60.7 -120.5 -31.5
8 U A.U8 162.7 160.7 -159.9 -163.8 -142.6 176.2
9 A A.A9 -140.6 -38.9 -159.3 -112.7 157.1 -105.5
10 g A.2MG10 27.8 -130.3 97.2 -130.1 134.8 -110.3
11 C A.C11 170.3 -135.8 -175.7 -136.7 -137.8 -119.9
12 U A.U12 159.9 -121.6 176.5 -130.6 -148.5 -101.4
13 C A.C13 178.1 -179.1 -166.8 176.7 -118.5 178.4
14 A A.A14 171.9 -146.5 172.1 -133.4 -179.7 -74.6
15 G A.G15 164.3 -177.9 -166.6 -161.0 -92.6 -101.8
16 u A.H2U16 -124.1 -77.5 -114.2 -108.3 -72.5 -127.0
17 u A.H2U17 -10.5 -64.3 7.7 -94.7 17.3 -125.4
18 G A.G18 -21.0 -167.4 45.3 -160.9 61.3 -124.2
19 G A.G19 -127.4 -43.3 -122.0 -72.9 -105.8 -7.8
20 G A.G20 165.3 -100.4 -160.4 -101.1 -177.9 -115.4
21 A A.A21 -78.3 152.7 -68.0 155.1 -61.1 154.8
22 G A.G22 159.5 167.6 156.6 178.8 157.1 -162.6
23 A A.A23 178.4 -141.8 -173.5 -141.2 -156.1 -112.0
24 G A.G24 163.7 -139.5 177.7 -137.6 -137.6 -103.8
25 C A.C25 161.4 -132.6 179.2 -131.0 -128.2 -89.0
26 g A.M2G26 173.0 -133.0 -167.7 -130.4 -106.9 -93.6
27 C A.C27 163.5 -142.3 -178.0 -141.5 -123.6 -105.6
28 C A.C28 157.5 -143.8 171.1 -144.3 -136.3 -125.5
29 A A.A29 163.5 -152.9 179.0 -150.8 -142.9 -124.7
30 G A.G30 178.3 -127.8 -167.7 -126.5 -128.2 -72.5
31 A A.A31 165.4 -133.9 -174.3 -131.0 -101.0 -93.9
32 c A.OMC32 164.5 -139.2 -175.9 -138.0 -122.3 -108.9
33 U A.U33 165.1 -114.0 177.8 -158.5 -141.1 138.3
34 g A.OMG34 27.3 -121.7 50.5 -123.7 22.7 -84.4
35 A A.A35 162.5 -127.7 -177.7 -128.5 -116.8 -113.4
36 A A.A36 164.9 -172.7 -174.4 -169.2 -142.3 -115.1
37 g A.YYG37 163.1 -135.2 174.1 -131.3 -119.8 -79.8
38 A A.A38 170.2 -133.9 -173.3 -129.0 -104.3 -105.5
39 P A.PSU39 174.0 -132.6 -168.6 -131.2 -127.5 -89.6
40 c A.5MC40 163.1 -148.5 -177.6 -149.3 -115.9 -131.7
41 U A.U41 169.4 -148.8 177.2 -144.0 -152.9 -120.5
42 G A.G42 171.2 -150.4 -171.5 -151.6 -133.9 -124.5
43 G A.G43 174.2 -151.6 -174.4 -150.0 -134.0 -124.5
44 A A.A44 173.2 -120.4 -171.8 -120.0 -133.3 -72.6
45 G A.G45 168.6 -141.6 -168.3 -128.4 -103.4 -133.4
46 g A.7MG46 -143.2 -107.3 -133.6 -149.6 -148.2 -162.7
47 U A.U47 -31.5 -56.8 4.8 -91.0 24.9 -110.7
48 C A.C48 -82.5 53.9 -29.3 17.5 1.5 -107.6
49 c A.5MC49 -56.7 -145.3 -36.6 -142.8 103.2 -130.2
50 U A.U50 174.8 -146.6 -176.9 -142.8 -153.6 -113.8
51 G A.G51 170.3 -147.3 -175.5 -148.2 -134.2 -122.1
52 U A.U52 160.3 -145.8 173.9 -144.3 -141.8 -119.6
53 G A.G53 174.9 -141.5 -167.2 -142.4 -124.7 -111.6
54 t A.5MU54 171.1 -129.2 -177.4 -122.6 -133.3 -76.4
55 P A.PSU55 165.3 -115.2 -173.6 -155.4 -112.1 145.1
56 C A.C56 31.4 -126.9 51.6 -124.1 25.3 -87.4
57 G A.G57 164.3 -142.5 -174.1 -131.9 -119.2 -113.8
58 a A.1MA58 -131.5 -108.7 -105.3 -171.2 -104.2 159.8
59 U A.U59 1.8 -119.4 26.8 -109.9 49.0 -56.9
60 C A.C60 -171.8 -40.7 -130.1 -68.5 -70.2 -35.8
61 C A.C61 122.4 -148.3 168.6 -144.1 -158.2 -117.4
62 A A.A62 173.0 -146.6 -176.9 -144.9 -142.0 -119.6
63 C A.C63 164.5 -148.3 177.9 -149.6 -143.9 -128.6
64 A A.A64 158.4 -151.0 168.5 -148.2 -154.8 -122.8
65 G A.G65 173.6 -147.3 -172.0 -145.4 -130.5 -121.2
66 A A.A66 177.6 -145.4 -170.1 -142.7 -133.5 -111.9
67 A A.A67 165.6 -149.3 -176.9 -149.8 -129.8 -126.7
68 U A.U68 168.9 -138.2 179.4 -136.1 -143.2 -96.5
69 U A.U69 165.6 -160.5 -176.0 -161.2 -118.8 -156.9
70 C A.C70 166.7 -146.2 173.6 -149.0 -171.6 -127.0
71 G A.G71 161.0 -143.0 174.0 -142.3 -146.3 -113.4
72 C A.C72 166.1 -141.5 -177.5 -141.9 -131.5 -110.2
73 A A.A73 167.6 -137.8 -177.2 -133.3 -127.1 -89.8
74 C A.C74 171.2 -122.1 -172.8 -116.5 -116.2 -72.1
75 C A.C75 174.9 106.5 -161.9 109.8 -102.9 -139.3
76 A A.A76 --- --- --- --- --- ---
******************************************************************************************
Sugar conformational parameters:
v0: C4'-O4'-C1'-C2'
v1: O4'-C1'-C2'-C3'
v2: C1'-C2'-C3'-C4'
v3: C2'-C3'-C4'-O4'
v4: C3'-C4'-O4'-C1'
tm: the amplitude of pucker
P: the phase angle of pseudorotation
[Ref: Altona & Sundaralingam (1972): "Conformational analysis
of the sugar ring in nucleosides and nucleotides. A new
description using the concept of pseudorotation."
J Am Chem Soc, 94(23):8205-8212]
nt v0 v1 v2 v3 v4 tm P Puckering
1 G A.G1 1.7 -23.4 35.1 -35.2 21.1 36.5 16.1 C3'-endo
2 C A.C2 1.6 -23.2 34.8 -34.8 20.9 36.2 16.1 C3'-endo
3 G A.G3 2.7 -25.1 36.8 -36.1 21.2 38.1 14.6 C3'-endo
4 G A.G4 -1.6 -22.3 36.3 -38.2 25.0 38.8 20.8 C3'-endo
5 A A.A5 10.1 -32.6 41.5 -36.6 16.7 41.7 4.8 C3'-endo
6 U A.U6 0.3 -24.0 37.3 -38.1 23.9 39.2 18.2 C3'-endo
7 U A.U7 -24.4 35.4 -32.4 18.9 3.3 35.4 156.1 C2'-endo
8 U A.U8 5.8 -28.7 39.7 -37.2 19.7 40.4 10.5 C3'-endo
9 A A.A9 -31.7 41.8 -35.6 18.1 8.4 41.2 149.8 C2'-endo
10 g A.2MG10 7.8 -28.0 36.7 -33.0 15.9 37.0 6.6 C3'-endo
11 C A.C11 1.2 -21.2 32.1 -32.5 19.8 33.5 16.8 C3'-endo
12 U A.U12 -4.6 -19.3 34.5 -37.9 26.7 38.1 25.2 C3'-endo
13 C A.C13 -3.4 -19.4 33.8 -36.4 25.1 36.9 23.7 C3'-endo
14 A A.A14 12.6 -30.8 36.8 -30.2 11.0 36.8 358.9 C2'-exo
15 G A.G15 1.9 -24.6 36.8 -36.8 22.2 38.3 16.0 C3'-endo
16 u A.H2U16 0.0 -18.7 29.2 -30.2 19.2 30.9 18.8 C3'-endo
17 u A.H2U17 23.0 -36.7 35.1 -23.2 0.2 37.0 341.4 C2'-exo
18 G A.G18 -27.9 39.5 -35.0 20.2 4.8 38.9 154.3 C2'-endo
19 G A.G19 -17.6 31.0 -31.9 23.1 -3.8 32.7 167.6 C2'-endo
20 G A.G20 6.6 -27.8 36.6 -34.2 17.5 37.0 8.7 C3'-endo
21 A A.A21 3.8 -25.0 35.1 -34.4 19.4 36.0 13.0 C3'-endo
22 G A.G22 16.4 -34.1 38.1 -29.5 8.3 38.3 353.8 C2'-exo
23 A A.A23 4.2 -26.6 37.4 -36.5 20.1 38.3 12.6 C3'-endo
24 G A.G24 3.9 -28.4 40.3 -39.3 22.4 41.5 13.4 C3'-endo
25 C A.C25 0.6 -24.5 37.8 -38.0 23.6 39.6 17.4 C3'-endo
26 g A.M2G26 6.3 -27.5 37.1 -34.7 17.9 37.6 9.3 C3'-endo
27 C A.C27 0.2 -23.5 36.5 -37.2 23.6 38.4 18.3 C3'-endo
28 C A.C28 6.6 -29.0 39.1 -36.3 18.8 39.6 9.2 C3'-endo
29 A A.A29 3.4 -26.6 38.4 -37.4 21.4 39.5 13.7 C3'-endo
30 G A.G30 2.6 -24.2 35.7 -34.9 20.4 36.9 14.5 C3'-endo
31 A A.A31 2.6 -24.0 35.0 -34.6 20.2 36.2 14.6 C3'-endo
32 c A.OMC32 -1.2 -21.7 35.1 -36.7 23.9 37.4 20.4 C3'-endo
33 U A.U33 3.5 -25.4 36.5 -35.3 20.1 37.5 13.3 C3'-endo
34 g A.OMG34 3.9 -22.7 32.2 -30.8 17.1 32.9 12.2 C3'-endo
35 A A.A35 -2.0 -19.9 32.7 -34.9 23.4 35.2 21.9 C3'-endo
36 A A.A36 -20.6 -7.3 30.6 -43.2 40.5 43.9 45.8 C4'-exo
37 g A.YYG37 2.1 -24.1 36.0 -35.6 21.0 37.4 15.4 C3'-endo
38 A A.A38 10.9 -30.3 37.6 -32.5 13.6 37.7 2.4 C3'-endo
39 P A.PSU39 2.1 -25.6 38.5 -38.4 22.8 40.0 15.8 C3'-endo
40 c A.5MC40 0.8 -22.5 34.6 -35.0 21.5 36.3 17.3 C3'-endo
41 U A.U41 3.8 -27.7 39.9 -38.6 22.0 41.0 13.4 C3'-endo
42 G A.G42 -1.7 -22.4 36.8 -38.6 25.4 39.4 20.9 C3'-endo
43 G A.G43 4.3 -27.6 39.1 -37.6 21.1 40.1 12.6 C3'-endo
44 A A.A44 1.0 -23.0 35.2 -35.4 21.6 36.8 16.9 C3'-endo
45 G A.G45 2.1 -24.3 35.7 -35.4 21.2 37.0 15.4 C3'-endo
46 g A.7MG46 -27.4 38.6 -34.7 19.7 4.7 38.5 154.5 C2'-endo
47 U A.U47 -20.9 34.8 -35.1 24.3 -2.2 36.4 164.9 C2'-endo
48 C A.C48 -25.6 38.4 -35.6 22.1 2.1 38.4 158.2 C2'-endo
49 c A.5MC49 5.8 -28.1 38.7 -36.0 19.1 39.3 10.1 C3'-endo
50 U A.U50 9.4 -32.2 41.0 -36.4 17.6 41.2 5.6 C3'-endo
51 G A.G51 4.9 -27.9 38.9 -36.8 20.3 39.7 11.5 C3'-endo
52 U A.U52 3.2 -28.5 41.4 -40.1 23.6 42.7 14.1 C3'-endo
53 G A.G53 -1.0 -23.1 37.0 -38.3 24.9 39.4 19.8 C3'-endo
54 t A.5MU54 -1.4 -22.2 35.9 -37.7 24.8 38.3 20.6 C3'-endo
55 P A.PSU55 6.2 -29.9 40.9 -38.3 20.4 41.5 10.1 C3'-endo
56 C A.C56 3.8 -25.3 35.7 -34.5 19.2 36.6 12.6 C3'-endo
57 G A.G57 4.0 -26.7 37.9 -36.5 20.6 38.9 12.8 C3'-endo
58 a A.1MA58 -24.3 38.4 -36.9 23.9 0.2 39.0 161.1 C2'-endo
59 U A.U59 -4.4 -18.3 31.8 -35.7 25.4 35.3 25.8 C3'-endo
60 C A.C60 -28.8 40.5 -36.4 21.2 4.7 40.3 154.8 C2'-endo
61 C A.C61 2.6 -25.5 36.8 -36.6 21.5 38.1 14.8 C3'-endo
62 A A.A62 5.9 -27.8 38.1 -35.4 18.8 38.7 9.9 C3'-endo
63 C A.C63 5.4 -27.3 37.5 -35.5 19.1 38.1 10.7 C3'-endo
64 A A.A64 4.1 -28.6 40.2 -38.8 22.2 41.2 12.9 C3'-endo
65 G A.G65 1.5 -26.6 39.5 -39.9 24.3 41.2 16.5 C3'-endo
66 A A.A66 -2.9 -21.6 36.5 -38.8 26.5 39.5 22.5 C3'-endo
67 A A.A67 2.4 -24.9 36.5 -36.1 21.4 37.8 15.0 C3'-endo
68 U A.U68 5.3 -28.4 39.5 -37.5 20.3 40.3 11.2 C3'-endo
69 U A.U69 2.9 -26.3 38.3 -37.9 22.3 39.6 14.7 C3'-endo
70 C A.C70 2.4 -25.9 38.7 -37.9 22.4 40.1 15.0 C3'-endo
71 G A.G71 3.7 -27.4 39.2 -38.3 21.8 40.3 13.5 C3'-endo
72 C A.C72 -0.6 -21.9 34.9 -36.2 23.1 37.0 19.5 C3'-endo
73 A A.A73 2.4 -25.4 37.3 -36.9 21.8 38.6 15.0 C3'-endo
74 C A.C74 4.4 -25.4 35.6 -34.0 18.6 36.4 11.8 C3'-endo
75 C A.C75 2.3 -22.5 33.1 -33.0 19.2 34.3 15.1 C3'-endo
76 A A.A76 -13.6 30.5 -34.8 27.7 -9.1 34.8 176.1 C2'-endo
******************************************************************************************
Assignment of sugar-phosphate backbone suites
bin: name of the 12 bins based on [delta(i-1), delta, gamma], where
delta(i-1) and delta can be either 3 (for C3'-endo sugar) or 2
(for C2'-endo) and gamma can be p/t/m (for gauche+/trans/gauche-
conformations, respectively) (2x2x3=12 combinations: 33p, 33t,
... 22m); 'inc' refers to incomplete cases (i.e., with missing
torsions), and 'trig' to triages (i.e., with torsion angle
outliers)
cluster: 2-char suite name, for one of 53 reported clusters (46
certain and 7 wannabes), '__' for incomplete cases, and
'!!' for outliers
suiteness: measure of conformer-match quality (low to high in range 0 to 1)
[Ref: Richardson et al. (2008): "RNA backbone: consensus all-angle
conformers and modular string nomenclature (an RNA Ontology
Consortium contribution)." RNA, 14(3):465-481]
nt bin cluster suiteness
1 G A.G1 inc __ 0
2 C A.C2 33p 1a 0.935
3 G A.G3 33p 1a 0.868
4 G A.G4 33p 1a 0.842
5 A A.A5 33p 1a 0.847
6 U A.U6 33p 1a 0.664
7 U A.U7 32p 1b 0.803
8 U A.U8 23p 2a 0.509
9 A A.A9 32p 1[ 0.046
10 g A.2MG10 23p 2g 0.640
11 C A.C11 33p 1a 0.507
12 U A.U12 33p 1a 0.898
13 C A.C13 33t 1c 0.824
14 A A.A14 trig !! 0
15 G A.G15 33p 1a 0.484
16 u A.H2U16 trig !! 0
17 u A.H2U17 33t !! 0
18 G A.G18 32p 5p 0.026
19 G A.G19 22p 4b 0.512
20 G A.G20 23p 2a 0.623
21 A A.A21 33t !! 0
22 G A.G22 33t 1f 0.714
23 A A.A23 33p 1a 0.840
24 G A.G24 33p 1a 0.881
25 C A.C25 33p 1a 0.967
26 g A.M2G26 33p 1a 0.819
27 C A.C27 33p 1a 0.698
28 C A.C28 33p 1a 0.923
29 A A.A29 33p 1a 0.973
30 G A.G30 33p 1a 0.838
31 A A.A31 33p 1a 0.914
32 c A.OMC32 33p 1a 0.782
33 U A.U33 33p 1a 0.897
34 g A.OMG34 33p 1g 0.784
35 A A.A35 33p 1a 0.517
36 A A.A36 33p 1a 0.670
37 g A.YYG37 33p 1a 0.625
38 A A.A38 33p 1a 0.903
39 P A.PSU39 33p 1a 0.680
40 c A.5MC40 33p 1a 0.942
41 U A.U41 33p 1a 0.945
42 G A.G42 33p 1a 0.630
43 G A.G43 33p 1a 0.882
44 A A.A44 33p 1a 0.837
45 G A.G45 33p 1a 0.749
46 g A.7MG46 32p 1[ 0.849
47 U A.U47 22p 4p 0.589
48 C A.C48 22t 2u 0.283
49 c A.5MC49 23p 6d 0.520
50 U A.U50 33p 1a 0.656
51 G A.G51 33p 1a 0.981
52 U A.U52 33p 1a 0.945
53 G A.G53 33p 1a 0.896
54 t A.5MU54 33p 1a 0.720
55 P A.PSU55 33p 1a 0.586
56 C A.C56 33p 1g 0.894
57 G A.G57 33p 1a 0.837
58 a A.1MA58 32p 1[ 0.332
59 U A.U59 23p 4d 0.411
60 C A.C60 32p 1b 0.662
61 C A.C61 23p 2a 0.553
62 A A.A62 33p 1a 0.895
63 C A.C63 33p 1a 0.964
64 A A.A64 33p 1a 0.791
65 G A.G65 33p 1a 0.586
66 A A.A66 33p 1a 0.940
67 A A.A67 33p 1a 0.941
68 U A.U68 33p 1a 0.891
69 U A.U69 33p 1a 0.951
70 C A.C70 33p 1a 0.809
71 G A.G71 33p 1a 0.761
72 C A.C72 33p 1a 0.832
73 A A.A73 33p 1a 0.965
74 C A.C74 33p 1a 0.886
75 C A.C75 33p 1a 0.639
76 A A.A76 32t !! 0
Concatenated suite string per chain. To avoid confusion of lower case
modified nucleotide name (e.g., 'a') with suite cluster (e.g., '1a'),
use --suite-delimiter to add delimiters (matched '()' by default).
1 A RNA nts=76 G1aC1aG1aG1aA1aU1bU2aU1[A2gg1aC1aU1cC!!A1aG!!u!!u5pG4bG2aG!!A1fG1aA1aG1aC1ag1aC1aC1aA1aG1aA1ac1aU1gg1aA1aA1ag1aA1aP1ac1aU1aG1aG1aA1aG1[g4pU2uC6dc1aU1aG1aU1aG1at1aP1gC1aG1[a4dU1bC2aC1aA1aC1aA1aG1aA1aA1aU1aU1aC1aG1aC1aA1aC1aC!!A
It gives me great pleasure to announce that the 3DNA/DSSR project is now funded by the NIH R24GM153869 grant, titled "X3DNA-DSSR: a resource for structural bioinformatics of nucleic acids". I am deeply grateful for the opportunity to continue working on a project that has basically defined who I am. It was a tough time during the funding gap over the past few years. Nevertheless, I have experienced and learned a lot, and witnessed miracles enabled by enthusiastic users.
Since late 2020 when I lost my R01 grant, DSSR has been licensed by the Columbia Technology Ventures (CTV). I appreciate the numerous users (including big pharma) who purchased a DSSR Pro License or a DSSR Basic paid License. Thanks to the NIH R24GM153869 grant, we are pleased to provide DSSR Basic free of charge to the academic community. Academic Users may submit a license request for DSSR Basic or DSSR Pro by clicking "Express Licensing" on the CTV landing page. Commercial users may inquire about pricing and licensing terms by emailing techtransfer@columbia.edu, copying xiangjun@x3dna.org.
DSSR v2.4.5-2024sep24 was released to synchronize with the new R24 funding, which will bring the project to an entirely new level. All existing users are encouraged to upgrade their installation to this release which contains miscellaneous bug fixes (e.g., chain id with > 4 chars) and numerous minor improvements.
Lots of exciting things will happen for the project. The first thing is to make DSSR freely accessible to the academic community. In the past couple of weeks, CTV have already issued quite a few DSSR Basic Academic licenses to users from all over the world. So the demand is high, and it will become stronger as more academic users become aware of DSSR. I'm closely monitoring the 3DNA Forum, and is always ready to answer users questions.
I am committed to making DSSR a brand that stands for quality and value. By virtue of its unmatched functionality, usability, and support, DSSR saves users a substantial amount of time and effort when compared to other options. My track record throughout the years has unambiguously demonstrated my dedication to this solid software product.
DSSR Basic contains all features described in the three DSSR-related papers, and includes the originally separate SNAP program (still unpublished) for analyzing DNA/RNA-protein complexes. The Pro version integrates the classic 3DNA functionality, plus advanced modeling routines, with email/Zoom/phone support.
The DSSR-PyMOL schematics have been featured in all 12 cover images (January to December) of the RNA Journal in 2021. Moreover, the January 2022 issue of RNA continues to highlight DSSR-enabled schematics (see the note below). In the current Covid-19 pandemic, this cover seems to be a fit for the upcoming Christmas holiday season.
Ebola virus matrix protein octameric ring (PDB id: 7K5L; Landeras-Bueno S, Wasserman H, Oliveira G, VanAernum ZL, Busch F, Salie ZL, Wysocki VH, Andersen K, Saphire EO. 2021. Cellular mRNA triggers structural transformation of Ebola virus matrix protein VP40 to its essential regulatory form. Cell Rep 35: 108986). The Ebola virus matrix protein (VP40) forms distinct structures linked to distinct functions in the virus life cycle. VP40 forms an octameric ring-shaped (D4 symmetry) assembly upon binding of RNA and is associated with transcriptional control. RNA backbone is displayed as a red ribbon; block bases use NDB colors: A—red, G—green, U—cyan; protein is displayed as a gold ribbon. Cover image provided by the Nucleic Acid Database (ndbserver.rutgers.edu). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
Thanks to Dr. Cathy Lawson at the NDB for generating these cover images using DSSR and PyMOL for the RNA Journal. I’m gratified that the 2020 NAR paper is explicitly acknowledged: it’s the first time I’ve published as a single author in my scientific career.

Did you know that you can easily generate similar DSSR-PyMOL schematics via the http://skmatic.x3dna.org/ website? It is “simple and effective”, “good for teaching”, and has been highly recommended by Dr. Quentin Vicens (CU Denver) in FacultyOpinions.com.
The 12 PDB structures illustrated in the 2021 covers are:
- January 2021 “iMango-III fluorescent aptamer (PDB id: 6PQ7; Trachman III RJ, Stagno JR, Conrad C, Jones CP, Fischer P, Meents A, Wang YX, Ferre-D’Amare AR. 2019. Co-crystal structure of the iMango-III fluorescent RNA aptamer using an X-ray free-electron laser. Acta Cryst F 75: 547). Upon binding TO1-biotin, the iMango-III aptamer achieves the largest fluorescence enhancement reported for turn-on aptamers (over 5000-fold).”
- February 2021 “Human adenosine deaminase (E488Q mutant) acting on dsRNA (PDB id: 6VFF; Thuy-Boun AS, Thomas JM, Grajo HL, Palumbo CM, Park S, Nguyen LT, Fisher AJ, Beal PA. 2020. Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa532). Adenosine deaminase enzymes convert adenosine to inosine in duplex RNA, a modification that strongly affects RNA structure and function in multiple ways.”
- March 2021 “Hepatitis A virus IRES domain V in complex with Fab (PDB id: 6MWN; Koirala D, Shao Y, Koldobskaya Y, Fuller JR, Watkins AM, Shelke SA, Pilipenko EV, Das R, Rice PA, Piccirilli JA. 2019. A conserved RNA structural motif for organizing topology within picornaviral internal ribosome entry sites. Nat Commun 10: 3629).”
- April 2021 “Mouse endonuclease V in complex with 23mer RNA (PDB id: 6OZO; Wu J, Samara NL, Kuraoka I, Yang W. 2019. Evolution of inosine-specific endonuclease V from bacterial DNase to eukaryotic RNase. Mol Cell 76: 44). Endonuclease V cleaves the second phosphodiester bond 3′ to a deaminated adenosine (inosine). Although highly conserved, EndoV change substrate preference from DNA in bacteria to RNA in eukaryotes.”
- May 2021 “Manganese riboswitch from Xanthmonas oryzae (PDB id: 6N2V; Suddala KC, Price IR, Dandpat SS, Janeček M, Kührová P, Šponer J, Banáš P, Ke A, Walter NG. 2019. Local-to-global signal transduction at the core of a Mn2+ sensing riboswitch. Nat Commun 10: 4304). Bacterial manganese riboswitches control the expression of Mn2+ homeostasis genes. Using FRET, it was shown that an extended 4-way-junction samples transient docked states in the presence of Mg2+ but can only dock stably upon addition of submillimolar Mn2+.”
- June 2021 “Sulfolobus islandicus Csx1 RNase in complex with cyclic RNA activator (PDB id: 6R9R; Molina R, Stella S, Feng M, Sofos N, Jauniskis V, Pozdnyakova I, Lopez-Mendez B, She Q, Montoya G. 2019. Structure of Csx1-cOA4 complex reveals the basis of RNA decay in Type III-B CRISPR-Cas. Nat Commun 10: 4302). CRISPR-Cas multisubunit complexes cleave ssRNA and ssDNA, promoting the generation of cyclic oligoadenylate (cOA), which activates associated CRISPR-Cas RNases. The Csx1 RNase dimer is shown with cyclic (A4) RNA bound.”
- July 2021 “M. tuberculosis ileS T-box riboswitch in complex with tRNA (PDB id: 6UFG; Battaglia RA, Grigg JC, Ke A. 2019. Structural basis for tRNA decoding and aminoacylation sensing by T-box riboregulators. Nat Struct Mol Biol 26: 1106). T-box riboregulators are a class of cis-regulatory RNAs that govern the bacterial response to amino acid starvation by binding, decoding, and reading the aminoacylation status of specific transfer RNAs.”
- August 2021 “CAG repeats recognized by cyclic mismatch binding ligand (PDB id: 6QIV; Mukherjee S, Blaszczyk L, Rypniewski W, Falschlunger C, Micura R, Murata A, Dohno C, Nakatan K, Kiliszek A. 2019. Structural insights into synthetic ligands targeting A–A pairs in disease-related CAG RNA repeats. Nucleic Acids Res 47:10906). A large number of hereditary neurodegenerative human diseases are associated with abnormal expansion of repeated sequences. RNA containing CAG repeats can be recognized by synthetic cyclic mismatch-binding ligands such as the structure shown.”
- September 2021 “Corn aptamer complex with fluorophore Thioflavin T (PDB id: 6E81; Sjekloca L, Ferre-D’Amare AR. 2019. Binding between G quadruplexes at the homodimer interface of the Corn RNA aptamer strongly activates Thioflavin T fluorescence. Cell Chem Biol 26: 1159). The fluorescent compound Thioflavin T, widely used for the detection of amyloids, is bound at the dimer interface of the homodimeric G-quadruplex-containing RNA Corn aptamer.”
- October 2021 “Cas9 nuclease-sgRNA complex with anti-CRISPR protein inhibitor (PDB id: 6JE9; Sun W, Yang J, Cheng Z, Amrani N, Liu C, Wang K, Ibraheim R, Edraki A, Huang X, Wang M, et al. 2019. Structures of Neisseria meningitidis Cas9 complexes in catalytically poised and anti-CRISPR-inhibited states. Mol Cell 76: 938–952.e5). Nme1Cas9, a compact nuclease for in vivo genome editing. AcrIIC3 is an anti-CRISPR protein inhibitor.”
- November 2021 “Two-quartet RNA parallel G-quadruplex complexed with porphyrin (PDB id: 6JJI; Zhang Y, Omari KE, Duman R, Liu S, Haider S, Wagner A, Parkinson GN, Wei D. 2020. Native de novo structural determinations of non-canonical nucleic acid motifs by X-ray crystallography at long wavelengths. Nucleic Acids Res 48: 9886–9898).”
- December 2021 “Structure of S. pombe Lsm1–7 with RNA, polyuridine with 3’ guanosine (PDB id: 6PPV; Montemayor EJ, Virta JM, Hayes SM, Nomura Y, Brow DA, Butcher SE. 2020. Molecular basis for the distinct cellular functions of the Lsm1–7 and Lsm2–8 complexes. RNA 26: 1400–1413). Eukaryotes possess eight highly conserved Lsm (like Sm) proteins that assemble into circular, heteroheptameric complexes, bind RNA, and direct a diverse range of biological processes. Among the many essential functions of Lsm proteins, the cytoplasmic Lsm1–7 complex initiates mRNA decay, while the nuclear Lsm2–8 complex acts as a chaperone for U6 spliceosomal RNA.”
On December 9, 2021, at 15:00 CET, I will present a BioExcel webinar titled “X3DNA-DSSR, a resource for structural bioinformatics of nucleic acids.”
For the record, the screenshot of the announcement is shown below:

Today, I released a video overview of DSSR (http://docs.x3dna.org/dssr-overview/).
DSSR has a sizable user base. However, in my opinion, DSSR is still underutilized for what it has to offer. This overview video is intended not only to attract new DSSR users, but also to highlight features that even experienced users may overlook.