
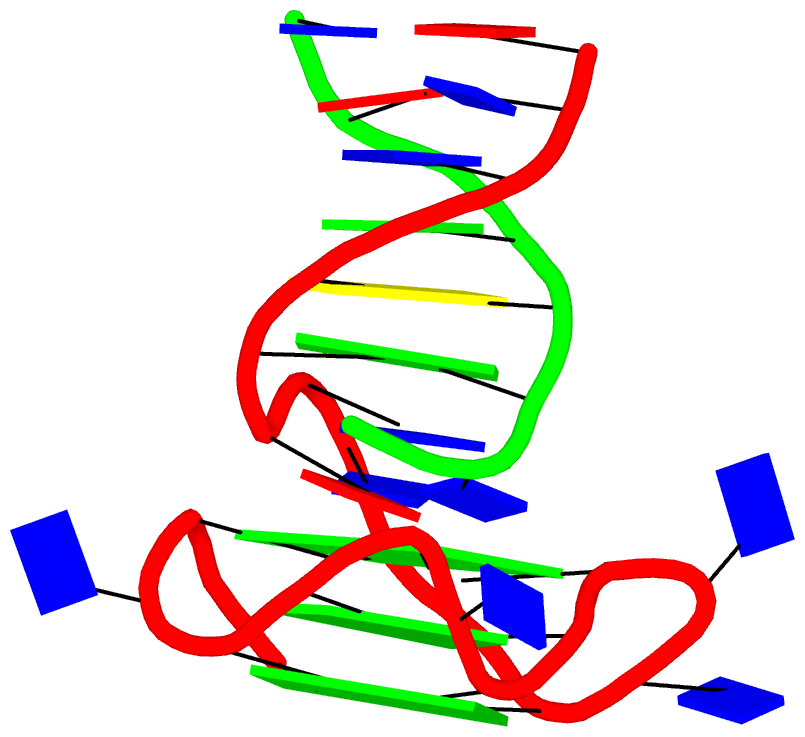
Crystal structure of SARS-CoV-2 stem–loop 5 (SL5) (PDB id: 9E9Q; Jones CP, Ferré-D'Amaré AR. 2025. Crystallographic and cryoEM analyses reveal SARS-CoV-2 SL5 is a mobile T-shaped four-way junction with deep pockets. RNA 31: 949–960). The T-shaped four-way junction of the coronavirus SL5 structural element provides a starting point for examining the structures of larger RNA motifs and their interactions with other molecules. Image highlighting the four arms of the junction. The RNA backbone is depicted by a gray ribbon. The bases within the arms of the junction are colored respectively in blue, red, yellow, and cyan. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for Structural Bioinformatics of Nucleic Acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
As the developer of DSSR, I am thrilled to see its application in cutting-edge research across multiple disciplines. Below is a list of four recent publications that highlight how DSSR has been utilized, underscoring its versatility and significance in structural bioinformatics.
In the Geng et al. (2025) Nucleic Acids Research (NAR) paper, titled 'Revealing hidden protonated conformational states in RNA dynamic ensembles', DSSR is simply cited as follows:
All bp geometries, hydrogen-bond, backbone, stacking, and sugar dihedral angles were calculated using X3DNA-DSSR [77].
In the preprint by Gordan et al. (2025), titled 'High-throughput characterization of transcription factors that modulate UV damage formation and repair at single-nucleotide resolution', DSSR is cited as follows:
Step base stacking, base pair shift, base pair slide, interbase angle, pseudorotation angle, and sugar puckering classifications of nucleobases were computed using X3DNA-DSSR (v2.5.0)75. Base stacking was defined as the overlapping polygon area in Å2 when projecting the dipyrimidine base ring atoms (excluding exocyclic atoms) into the mean base pair plane76. The sugar ring pseudorotation phase angle of each pyrimidine was also calculated using X3DNA-DSSR as described by Altona, C. & Sundaralingam, M.77 Interbase angle was defined as sqrt(propeller2+buckle2) per the X3DNA-DSSR documentation.
Figure 6: TF Binding Induces Structural Distortion Favorable to UV Dimerization is highly informative, particularly panel (a), which illustrates the ensemble of structural parameters that predispose dipyrimidines to cyclobutane pyrimidine dimers (CPD) or 6-4 pyrimidine-pyrimidones (6-4 PP) formation. DSSR is designed as an integrated software tool, offering a comprehensive suite of structural parameters not found in any other single tool I am aware of. Despite this, the innovative use of DSSR by Gordan et al. exceeds my expectations and demonstrates its versatility.
In the preprint by Kubaney et al. (2025) from the Baker group, titled 'RNA sequence design and protein-DNA specificity prediction with NA-MPNN', DSSR is cited as follows:
On the pseudoknot subset, we evaluate additional structure‐ and reactivity‐based metrics. DSSR v2.3.241 is used to extract the ground‐truth secondary structure from the native crystal structures. For each designed sequence, RibonanzaNet predicts 2A3 reactivity profiles, from which we compute predicted OpenKnot scores (see https://github.com/eternagame/OpenKnotScore)31 using the predicted reactivity together with the DSSR ground truth.
In a recent NSMB paper from the Baker group, titled 'Computational design of sequence-specific DNA-binding proteins', 3DNA is cited as follows:
RIF docking of scaffolds onto DNA targets (DBP design step 1) Structures of B-DNA for each target (Supplementary Table 2) were generated by (1) using the DNA portion of PDB 1BC8 (ref. 60), PDB 1YO5 (ref. 61), PDB 1L3L (ref. 51) or PDB 2O4A (ref. 62) or (2) using the software X3DNA63, followed by a constrained Rosetta relax of the DNA structure.
Please note that 3DNA has been replaced by DSSR. The functionality for constructing B-DNA models, previously provided by 3DNA, is now directly available in DSSR via its fiber and rebuild modules.
In the preprint by Si et al. (2025), titled 'End-to-End Single-Stranded DNA Sequence Design with All-Atom Structure Reconstruction', DSSR is cited as follows:
Since ViennaRNA and NUPACK require secondary structures as input, we used DSSR35 to extract secondary structures from the corresponding ssDNA three-dimensional structures.
The above use cases are merely a sample of how DSSR is utilized in the scientific literature. It is reasonable to state that DSSR has emerged as a de facto standard tool within the field of nucleic acid structural bioinformatics. Overall, DSSR is a mature, robust, and efficient software product that is actively developed and maintained. I am committed to making DSSR synonymous with quality and value. Its unmatched functionality, usability, and support save users significant time and effort compared to alternative solutions.
DSSR is available free of charge for academic users. Additionally, it has been integrated into other high-profile bioinformatics resources, including NAKB, PDB-redo, and N•ESPript.
References
- Geng A, Roy R, Ganser L, Li L, Al-Hashimi HM. Revealing hidden protonated conformational states in RNA dynamic ensembles. Nucleic Acids Research. 2025;53:gkaf1366. https://doi.org/10.1093/nar/gkaf1366.
- Gordan R, Wasserman H, Chi B, Bohm K, Duan M, Sahay H, et al. High-throughput characterization of transcription factors that modulate UV damage formation and repair at single-nucleotide resolution. 2025. https://doi.org/10.21203/rs.3.rs-8197218/v1.
- Kubaney A, Favor A, McHugh L, Mitra R, Pecoraro R, Dauparas J, et al. RNA sequence design and protein–DNA specificity prediction with NA-MPNN. 2025. https://doi.org/10.1101/2025.10.03.679414.
- Glasscock CJ, Pecoraro RJ, McHugh R, Doyle LA, Chen W, Boivin O, et al. Computational design of sequence-specific DNA-binding proteins. Nat Struct Mol Biol. 2025;32:2252–61. https://doi.org/10.1038/s41594-025-01669-4.
- Si Y, Xu Y, Chen L. End-to-end single-stranded DNA sequence design with all-atom structure reconstruction. 2025. https://doi.org/10.64898/2025.12.05.692525.
The DSSR-Jmol paper, titled "DSSR-enhanced visualization of nucleic acid structures in Jmol", has been officially published in the 2017 web-server issue of Nucleic Acids Research (NAR). Notably, the work has been featured in the cover image, as shown below:

Caption: 3D interactive visualization of selected RNA structural features enabled by the DSSR-Jmol integration (http://jmol.x3dna.org). Clockwise from upper left: Structure of the xpt-pbuX guanine riboswitch in complex with hypoxanthine (PDB id: 4fe5) in ‘base blocks’ representation. The three-way junction loop encompassing the metabolite (in space-filling representation) is color-coded by base identity: A, red; C, yellow; G, green; U, cyan. The loop-loop interaction (a kissing-loop motif) at the top is highlighted in red (upper left corner). Structure of the Thermus thermophilus 30S ribosomal subunit in complex with antibiotics (PDB id: 1fjg) in step diagram. The 16S ribosomal RNA is color-coded in spectrum with the 5′-end in blue and the 3′-end in red (upper middle). Structure of the classic L-shaped yeast phenylalanine tRNA (PDB id: 1ehz) in step diagram, with the three hairpin loops highlighted in red and the [2,1,5,0] four-way junction loop in blue (upper right corner). Structure of the Pistol self-cleaving ribozyme (PDB id: 5ktj), showcasing (in red) the horizontal helix in space-filling representation. The helix is composed of six short stems stabilized via coaxial stacking interactions (bottom).
The DSSR-Jmol integration bridges the DSSR command-line analyzing tool and the Jmol molecular viewer seamlessly together via the standard JSON interface. Now users can select DSSR-derived RNA structural features (such as base pairs, double helices, various loops, etc.) and visualize them in novel representations in Jmol interactively. Moreover, fine-grained characteristics of these features can be queried via the Jmol SQL for DSSR. The DSSR-Jmol integration fills a gap in RNA structural bioinformatics, and brings RNA visualization to an entirely new level. The web interface (http://jmol.x3dna.org) is fully functional and easy to use, serving a huge user base of researchers, educators, and students alike.
Featured as the cover image of the 2017 NAR web-server issue, DSSR's publicity would surely increase through the DSSR-Jmol integration. Additionally, I've written a new post (on the 3DNA Forum) that provides the scripts and datafiles used to create the cover image.

I am pleased to announce the (advance online, May 3, 2017) publication of a new paper titled "DSSR-enhanced visualization of nucleic acid structures in Jmol" in Nucleic Acids Research (NAR). Co-authored by Robert Hanson (Jmol) and me (DSSR), the article will appear in the July 2017 web-server issue of NAR. Here are the key links related to the paper:
The DSSR-Jmol integration project was initiated in October 2013 when I approached Bob at a meeting organized by RCSB PDB at Rutgers. Thereafter, we met only once in July 2014 in Paris. Over the years, we have mostly communicated via email, occasionally facilitated by Skype. Our work bridges the DSSR command-line analyzing tool and the Jmol molecular viewer together via a simple JSON interface and a powerful query language. Users can now select DSSR-derived RNA structural features (such as base pairs, double helices, and various loops) as easily as they can select protein alpha-helices and beta-strands. Moreover, fine-grained characteristics of these features can be queried via Jmol SQL for DSSR (see examples below). Notably, the novel representation styles (step diagram and base blocks) and coloring schemes bring RNA visualization to an entirely new level (see Figure 3 of the paper).
load =1ehz/dssr # load yeast phenylalanine tRNA to Jmol with DSSR annotation
SELECT hairpins # select the three hairpin loops
SELECT junctions # select the four-way junction loop
select within(dssr, "nts WHERE is_modified") # select modified nucleotides (14 total)
SELECT within(dssr, "pairs WHERE name != 'WC'") # select non-Watson-Crick pairs
SELECT within(dssr, "pairs WHERE name = 'WC' OR name = 'Wobble'") # select canonical pairs
Select within(dssr, "pairs WHERE name != 'WC' AND name != 'Wobble'") # select non-canonical pairs
SELECT within(dssr, "pairs WHERE LW = 'tSW'") # select pairs of type tSW per Leontis-Westhof
The DSSR-Jmol integration fills a gap in RNA structural bioinformatics, serving a huge user base of researchers, educators, and students alike. Its functionality is freely accessible either via the Jmol application, or the JSmol-based website (http://jmol.x3dna.org). By adhering to web standards, the website is fully functional in all modern browsers on various computer/operating systems (including handheld devices, such as tablets and smart phones). The web interface is simple and intuitive, and new users can get started easily. It also allows power users to take full advantage of Jmol scripting via a command-line console.
This work also provides an example for integrating DSSR-derived features into other molecular graphics programs or bioinformatics pipelines involving nucleic acid structures. By design, DSSR is a stand-alone, command-line program written in ANSI C. The binary executables are only ~1MB in size, and self-contained. With zero dependencies, no setup or configuration, it is trivial to get DSSR up and running. DSSR uncovers a wide range of RNA/DNA structural features in a consistent, easily accessible framework. It possesses a much richer set of functionalities for nucleic acid structural analysis (see the DSSR User Manual) than any other existing tools I am aware of. Moreover, the program is efficient and robust, making it an ideal component to be integrated into other pipelines, especially via the standard and structured JSON interface.
Collaborating with Bob has been a truly exciting experience. The NAR-web publication represents a gratifying intermediate result along an on-going journey. Hopefully, others (may be some of you) can join us in pushing forward the field of RNA structural bioinformatics.
Recently, while analyzing a representative set of RNA structures from the PDB, I came across three weird entries. They are documented below, primarily for my own record.
- 5els — “Structure of the KH domain of T-STAR in complex with AAAUAA RNA”. There are two alternative conformations for the six-nt
AAAUAA RNA component, labeled A and B, respectively. Normally, the A/B alternative coordinates for each atom are put directly next to each other, and assigned the same chain id, as in 1msy for the phosphate group of G2669 on chain A. In 5els, however, the two alternative conformations (A/B) are separated into two chains: chain H for A, and chain I for B.
- 1vql — “The structure of the transition state analogue ‘DCSN’ bound to the large ribosomal subunit of Haloarcula marismortui”. The three-nt fragment DA179—C180—C181 on chain 4 is in the 3’—>5’ direction.
- 4r3i — “The crystal structure of m(6)A RNA with the YTHDC1 YTH domain”. The mmCIF file has a model number of 0, instead of 1 (as in other cases I am aware of).
Dear 3DNA Forum subscribers,
Here are some highlights of recent developments of 3DNA/DSSR:
Note: If you’ve difficulty in accessing the 3DNA homepage, possibly the case from mainland China (as I know it), please visit its duplicate at http://home.x3dna.org. This newsletter is written in Markdown, with a translated HTML version posted on the 3DNA homepage.
3DNA v2.3
The C source code is now available. Since the programs are written in strict ANSI C, 3DNA can be compiled (as is) on any computers/operating systems with a C (or C++) compiler. For user convenience, three binary distributions (with source code under the src/ subdirectory) are provided for Windows, Linux, and Mac OS X. The distributed Windows version works in native Windows (7 and up, via the cmd command-line interface, or ConEMU), MinGW/Msys (Msys2), and Cygwin, in either 32 or 64-bit.
A new set of ‘simple’ base-pair and step parameters was introduced to give ‘intuitive’ numerical values for non-Watson-Crick base pairs and associated steps. See the short communication titled Characterization of base pair geometry in the January 2016 issue of Computational Crystallography Newsletter (CCN).
The fiber program includes a new option, --pauling, for easy generation of Pauling & Corey triplex models of DNA/RNA with arbitrary base sequence. See my blogpost titled Pauling’s triplex model of nucleic acids is available in 3DNA.
Thomas Holder (PyMOL Principal Developer at Schrödinger, Inc.) has built a PyMOL wrapper to 3DNA fiber models. Now generating standard, regular DNA/RNA models in PyMOL is straightforward — thanks, Thomas!
DSSR (Dissecting the Spatial Structure of RNA)
Selected features of DSSR have been incorporated into Jmol (in collaboration with Robert Hanson, Jmol Principal Developer), and PyMOL (in collaboration with Thomas Holder). In Jmol application (via the Console window), one can now, for example, load =1ehz/dssr and then select hairpins; color red to see where the three hairpin loops are in 3D. The Jmol-DSSR web interface makes DSSR-enhanced visualization of nucleic acid structures in Jmol readily accessible to a broad user base, and has been employed in classes for educational purpose. A sample image of DSSR-derived cartoon-block representation via PyMOL is available for PDB entry 5dww, which has a G-quadruplex-duplex interface.
Since the publication of the Nucleic Acids Research paper in 2015, DSSR has been continuously refined and expanded, with a total of 36 new releases (from v1.2.8 to v1.6.4) as of this writing. Notably, the --json option provides DSSR-derived parameters in the simple, structured, and standard JSON format that can be easily parsed. This JSON output format is the (preferred) way for the outside world to interface with DSSR, and the Jmol-DSSR integration is built upon it. The --nmr option allows for batch processing of MODEL/ENDMDL-delineated NMR ensembles or trajectories of molecular dynamics (MD) simulations. Did you know that scripts and data files for reproducing the reported results are available in the DSSR-NAR paper section on the 3DNA Forum?
The User Manual is now 88-page long, covering nevertheless only the most common use cases of what DSSR has to offer. Miss a feature that you would like to have? Maybe it is already there or can be easily implemented in DSSR. Simply ask (on the 3DNA Forum), and I’ll try my best to help.
SNAP (Structures of Nucleic Acid-Protein complexes)
- SNAP aims to consolidate, refine, and significantly extend commonly used functionalities for DNA/RNA-protein structural analysis in one easy-to-use program. Currently in beta testing, SNAP is already fully functional, with features for characterizing the protein-nucleic acid interface and identifying amino acid-base pairing and stacking interactions.
A note for 3DNA/DSSR users in mainland China: It’s a pleasure to see the ~100 registrations on the 3DNA Forum with emails ending in .cn, 163.com, or qq.com etc., mostly from recent years. I’m planning a trip to China in 2017, and I’d be happy to meet some of you for academic exchanges and possible collaborations (学术交流、合作). If you’re interested, let’s get in touch!
Best regards,
Xiang-Jun
—
Dr. Xiang-Jun Lu (律祥俊)
Email: xiangjun@x3dna.org
Web: http://home.x3dna.org/
Forum: http://forum.x3dna.org/
In 1953, Pauling and Corey published an influential paper, titled A proposed structure for the nucleic acids, in Proc. Natl. Acad. Sci. (PNAS). Key features of the proposed model is summarized in their Letter to Nature, Structure of the Nucleic Acids, published in Nature on February 21, 1953.
We have formulated a structure for the nucleic acids which is compatible with the main features of the X-ray diagram and with the general principles of molecular structure, and which accounts satisfactorily for some of the chemical properties of the substances. The structure involves three intertwined helical polynucleotide chains. Each chain, which is formed by phosphate di-ester groups and linking β-D-ribofuranose or β-D-deoxyribofuranose residues with 3′, 5′ linkages, has approximately twenty-four nucleotide residues in seven turns of the helix. The helixes have the sense of a right-handed screw. The phosphate groups are closely packed about the axis of the molecule, with the pentose residues surrounding them, and the purine and pyrimidine groups projecting radially, their planes being approximately perpendicular to the molecular axis. The operation that converts one residue to the next residue in the polynucleotide chain is rotation by about 105° and translation by 3.4 Å.
This triplex model of nucleic acids, with phosphates in the center and bases on the outside, turned out to be fundamentally flawed. Yet, it played a significant role by prompting Watson and Crick in their discovery of the DNA double helix structure. While I’ve been aware of the Pauling triplex model from long ago, I had not read the original Pauling & Corey PNAS paper. Not surprisingly, I did not know what the triplex structure really looks like, other than some general ideas.
In a recent trip to Rutgers, Dr. Wilma Olson and I discussed the applications of fiber models collected in 3DNA. She drew my attention to the Pauling triplex model, and showed me Table 1 of the PNAS paper (see below), where the atomic coordinates for a nucleic acid repeating unit are listed.

The cylindrical format is the same as that for the fiber models in 3DNA. It thus seems fitting to add this historically significant triplex model to the collection. Googling revealed many interesting historical notes and comments, e.g. The Pauling-Corey Structure of DNA, and a short video Linus Pauling’s triple DNA helix model, 3D animation with basic narration. However, I failed to find a program that I can use to generate such a triplex model with generic base sequence. I decided to add the fiber --pauling option so users can easily create such a triplex model in 3D, just as they do for a classic A- and B-DNA duplex. This process has turned out to be very educational (detailed below), and the end result should be of general interest.
 in Pauling triplex")
- The left 3D image shows the nomenclature of atoms used by Pauling & Corey (see Table 1 above), which is dramatically different from current conventions. As an example, it should be the N1 atom of cytosine (a pyrimidine base), not N3, that is connected to the sugar C1′ atom in nowadays nomenclature. The corrections apply not only to base atoms, but also to the sugar and phosphate groups. The revised atom labeling (as used in the PDB) is illustrated in the 3D image on the right.
- Table 1 corresponds to the ribose sugar since it contains an O2′ atom (see also the figure above). The triplex model constructed would be RNA, but can be ‘converted’ to DNA by simply removing the O2′ atom (see below).
- Only the atomic coordinates for cytosine are listed in Table 1. The 3DNA
mutate_bases program came handy to get the corresponding atomic coordinates for A, G, T, and U. This expansion allows for the generation of Pauling’s triplex models with an arbitrary combination of the five common bases (A, C, G, T, and U).
- With the new
fiber --pauling option, now users can conveniently generate a Pauling’s triplex RNA/DNA model as shown below. Note that the one dash variant -pauling also works fine, with the additional -dna for DNA deoxyribose sugar. The PDB file (Pauling-triplex-mixed.pdb) with mixed DNA sequences can be downloaded, and the corresponding 3D image in top and side views is shown in the following figure.
fiber -pauling triplex-C10C10C10.pdb # default: 10 Cs per strand
fiber -pauling -seq=AAA triplex-A3A3A3.pdb # 3 As per strand
fiber -pauling -seq=AAAA:CCCC:GGGG Pauling-triplex-A4C4G4.pdb
fiber -pauling -seq=ACGGUU,UUGGAC,GGAACC Pauling-triplex-mixed.pdb
fiber --pauling-dna -seq=ACGGTT,TTGGAC,GGAACC Pauling-triplex-DNA.pdb

- With 3DNA’s
find_pair/analyze pair of programs, one can get the structural parameters corresponding to the Pauling triplex model. Not surprising, the repeating dinucleotide along each strand has a twist of 105°, and a rise of 3.4 Å. Notably, the sugar has a C2′-endo conformation.
3DNA contains 55 fiber models compiled from literature, plus a derived RNA model (as of v2.1). To the best of my knowledge, this is the most comprehensive collection of regular DNA/RNA models. Please see Table 4 of the 2003 3DNA NAR paper for detailed structural features of these models and references.
The 55 models are based on the following works:
- Chandrasekaran & Arnott (from #1 to #43) — the most well-known set of fiber models
- Alexeev et al. (#44-#45)
- van Dam & Levitt (#46-#47)
- Premilat & Albiser (#48-#55)
The utility program fiber makes the generation of all these fiber models in a simple, consistent interface, and produces coordinate files in either PDB or PDBML format. Of those models, some can be built with an arbitrary sequence of A, C, G and T (e.g., A-/B-/C-DNA from calf thymus), while others are of fixed sequences (e.g., Z-DNA with GC repeats). The sequence can be specified either from command-line or a plain text file, in either lower, UPPER, or MixED cases.
Once 3DNA in properly installed, the command-line interface is the most versatile and convenient way to generate, e.g., a regular double-stranded DNA (mostly, B-DNA) of arbitrary sequence. The command-help message (generated with fiber -h) is as below:
NAME
fiber - generate 55 fiber models based on Arnott and other's work
SYNOPSIS
fiber [OPTION] PDBFILE
DESCRIPTION
generate 55 fiber models based on the repeating unit from Arnott's
work, including the canonical A-, B-, C- and Z-DNA, triplex, etc
-xml output structure coordinates in PDBML format
-num a structure identification number in the range (1-55)
-m, -l brief description of the 55 fiber structures
-a, -1 A-DNA model (calf thymus)
-b, -4 B-DNA (calf thymus, default)
-c, -47 C-DNA (BII-type nucleotides)
-d, -48 D(A)-DNA ploy d(AT) : ploy d(AT) (right-handed)
-z, -15 Z-DNA poly d(GC) : poly d(GC)
-rna for RNA with arbitrary base sequence
-seq=string specifying an arbitrary base sequence
-single output a single-stranded structure
-h this help message (any non-recognized options will do)
INPUT
An structural identification number (symbol)
EXAMPLES
fiber fiber-BDNA.pdb
# fiber -4 fiber-BDNA.pdb
# fiber -b fiber-BDNA.pdb
fiber -a fiber-ADNA.pdb
fiber -seq=AAAGGUUU -rna fiber-RNA.pdb
fiber -seq=AAAGGUUU -rna -single fiber-ssRNA.pdb
OUTPUT
PDB file
SEE ALSO
analyze, anyhelix, find_pair
AUTHOR
3DNA v2.3-2016sept06, created and maintained by Xiang-Jun Lu (PhD)
Please post questions/comments on the 3DNA Forum: http://forum.x3dna.org/
Moreover, the w3DNA, 3D-DART web-interfaces, and the PyMOL wrapper make it easy to generate a regular DNA (or RNA) model, especially for occasional users or for educational purposes.
In principle, nothing is worth showing off with regard to 3DNA’s fiber model generation functionality. Nevertheless, this handy tool serves as a clear example of the differences between a “proof of concept” and a pragmatic software application. I initially decided to work on this tool simply for my own convenience. At that time, I had access to A-DNA and B-DNA fiber model generators, each as a separate program. Moreover, the constructed models did not comply to the PDB format in atom naming, among other subtitles.
I started with the Chandrasekaran & Arnott fiber models which I had a copy of data files. However, there were many details to work out, typos to correct, etc. to put them in a consistent framework. For other models, I had to read each original publication, and to type raw atomic cylindrical coordinates into computer. Again, quite a few inconsistencies popped up between the different publications with a time span over decades.
Overall, it was a quite tedious undertaking, requiring great attention to details. I am glad that I did that: I learned so much from the process, and more importantly, others can benefit from my effort. As I put in the 3DNA Nature Protocol paper (BOX 6 | FIBER-DIFFRACTION MODELS),
In preparing this set of fiber models, we have taken great care to ensure the accuracy and consistency of the models. For completeness and user verification, 3DNA includes, in addition to 3DNA-processed files, the original coordinates collected from the literature.
For those who want to understand what’s going on under the hood, there is no better way than to try to reproduce the process using, e.g., fiber B-DNA as an example.
From the very beginning, I had expected the 3DNA fiber functionality to serve as a handy tool for building a regular DNA duplex of chosen sequence. Over the years, the fiber program has gradually attracted attention from the community. The recent PyMOL wrapper by Thomas Holder is a clear sign of its increased popularity, and has prompted me to write this post, adapted largely from the one titled Fiber models in 3DNA make it easy to build regular DNA helices (dated Friday, October 9, 2009).
See also PyMOL wrapper to 3DNA fiber models
Given below is the content of the README file for fiber models in 3DNA:
1. The repeating units of each fiber structure are mostly based on the
work of Chandrasekaran & Arnott (from #1 to #43). More recent fiber
models are based on Alexeev et al. (#44-#45), van Dam & Levitt (#46
-#47) and Premilat & Albiser (#48-#55).
2. Clean up of each residue
a. currently ignore hydrogen atoms [can be easily added]
b. change ME/C7 group of thymine to C5M
c. re-assign O3' atom to be attached with C3'
d. change distance unit from nm to A [most of the entries]
e. re-ordering atoms according to the NDB convention
3. Fix up of problem structures.
a. str#8 has no N9 atom for guanine
b. str#10 is not available from the disk, manually input
c. str#14 C5M atom was named C5 for Thymine, resulting two C5 atoms
d. str#17 has wrong assignment of O3' atom on Guanine
e. str#33 has wrong C6 position in U3
f. str#37 to #str41 were typed in manually following Arnott's
new list as given in "Oxford Handbook of Nucleic Acid Structure"
edited by S. Neidle (Oxford Press, 1999)
g. str#38 coordinates for N6(A) and N3(T) are WRONG as given in the
original literature
h. str#39 and #40 have the same O3' coordinates for the 2nd strand
4. str#44 & 45 have fixed strand II residues (T)
5. str#46 & 47 have +z-axis upwards (based on BI.pdb & BII.pdb)
6. str#48 to 55 have +z-axis upwards
List of 55 fiber structures
id# Twist Rise Structure description
(dgrees) (A)
-------------------------------------------------------------------------------
1 32.7 2.548 A-DNA (calf thymus; generic sequence: A, C, G and T)
2 65.5 5.095 A-DNA poly d(ABr5U) : poly d(ABr5U)
3 0.0 28.030 A-DNA (calf thymus) poly d(A1T2C3G4G5A6A7T8G9G10T11) :
poly d(A1C2C3A4T5T6C7C8G9A10T11)
4 36.0 3.375 B-DNA (calf thymus; generic sequence: A, C, G and T)
5 72.0 6.720 B-DNA poly d(CG) : poly d(CG)
6 180.0 16.864 B-DNA (calf thymus) poly d(C1C2C3C4C5) : poly d(G6G7G8G9G10)
7 38.6 3.310 C-DNA (calf thymus; generic sequence: A, C, G and T)
8 40.0 3.312 C-DNA poly d(GGT) : poly d(ACC)
9 120.0 9.937 C-DNA poly d(G1G2T3) : poly d(A4C5C6)
10 80.0 6.467 C-DNA poly d(AG) : poly d(CT)
11 80.0 6.467 C-DNA poly d(A1G2) : poly d(C3T4)
12 45.0 3.013 D-DNA poly d(AAT) : poly d(ATT)
13 90.0 6.125 D-DNA poly d(CI) : poly d(CI)
14 -90.0 18.500 D-DNA poly d(A1T2A3T4A5T6) : poly d(A1T2A3T4A5T6)
15 -60.0 7.250 Z-DNA poly d(GC) : poly d(GC)
16 -51.4 7.571 Z-DNA poly d(As4T) : poly d(As4T)
17 0.0 10.200 L-DNA (calf thymus) poly d(GC) : poly d(GC)
18 36.0 3.230 B'-DNA alpha poly d(A) : poly d(T) (H-DNA)
19 36.0 3.233 B'-DNA beta2 poly d(A) : poly d(T) (H-DNA beta)
20 32.7 2.812 A-RNA poly (A) : poly (U)
21 30.0 3.000 A'-RNA poly (I) : poly (C)
22 32.7 2.560 Hybrid poly (A) : poly d(T)
23 32.0 2.780 Hybrid poly d(G) : poly (C)
24 36.0 3.130 Hybrid poly d(I) : poly (C)
25 32.7 3.060 Hybrid poly d(A) : poly (U)
26 36.0 3.010 10-fold poly (X) : poly (X)
27 32.7 2.518 11-fold poly (X) : poly (X)
28 32.7 2.596 Poly (s2U) : poly (s2U) (symmetric base-pair)
29 32.7 2.596 Poly (s2U) : poly (s2U) (asymmetric base-pair)
30 32.7 3.160 Poly d(C) : poly d(I) : poly d(C)
31 30.0 3.260 Poly d(T) : poly d(A) : poly d(T)
32 32.7 3.040 Poly (U) : poly (A) : poly(U) (11-fold)
33 30.0 3.040 Poly (U) : poly (A) : poly(U) (12-fold)
34 30.0 3.290 Poly (I) : poly (A) : poly(I)
35 31.3 3.410 Poly (I) : poly (I) : poly(I) : poly(I)
36 60.0 3.155 Poly (C) or poly (mC) or poly (eC)
37 36.0 3.200 B'-DNA beta2 Poly d(A) : poly d(U)
38 36.0 3.240 B'-DNA beta1 Poly d(A) : poly d(T)
39 72.0 6.480 B'-DNA beta2 Poly d(AI) : poly d(CT)
40 72.0 6.460 B'-DNA beta1 Poly d(AI) : poly d(CT)
41 144.0 13.540 B'-DNA Poly d(AATT) : poly d(AATT)
42 32.7 3.040 Poly(U) : poly d(A) : poly(U) [cf. #32]
43 36.0 3.200 Beta Poly d(A) : Poly d(U) [cf. #37]
44 36.0 3.233 Poly d(A) : poly d(T) (Ca salt)
45 36.0 3.233 Poly d(A) : poly d(T) (Na salt)
46 36.0 3.38 B-DNA (BI-type nucleotides; generic sequence: A, C, G and T)
47 40.0 3.32 C-DNA (BII-type nucleotides; generic sequence: A, C, G and T)
48 87.8 6.02 D(A)-DNA ploy d(AT) : ploy d(AT) (right-handed)
49 60.0 7.20 S-DNA ploy d(CG) : poly d(CG) (C_BG_A, right-handed)
50 60.0 7.20 S-DNA ploy d(GC) : poly d(GC) (C_AG_B, right-handed)
51 31.6 3.22 B*-DNA poly d(A) : poly d(T)
52 90.0 6.06 D(B)-DNA poly d(AT) : poly d(AT) [cf. #48]
53 -38.7 3.29 C-DNA (generic sequence: A, C, G and T) (depreciated)
54 32.73 2.56 A-DNA (generic sequence: A, C, G and T) [cf. #1]
55 36.0 3.39 B-DNA (generic sequence: A, C, G and T) [cf. #4]
-------------------------------------------------------------------------------
List 1-41 based on Struther Arnott: ``Polynucleotide secondary structures:
an historical perspective'', pp. 1-38 in ``Oxford Handbook of Nucleic
Acid Structure'' edited by Stephen Neidle (Oxford Press, 1999).
#42 and #43 are from Chandrasekaran & Arnott: "The Structures of DNA
and RNA Helices in Oriented Fibers", pp 31-170 in "Landolt-Bornstein
Numerical Data and Functional Relationships in Science and Technology"
edited by W. Saenger (Springer-Verlag, 1990).
#44-#45 based on Alexeev et al., ``The structure of poly(dA) . poly(dT)
as revealed by an X-ray fiber diffraction''. J. Biomol. Str. Dyn, 4,
pp. 989-1011, 1987.
#46-#47 based on van Dam & Levitt, ``BII nucleotides in the B and C forms
of natural-sequence polymeric DNA: a new model for the C form of DNA''.
J. Mol. Biol., 304, pp. 541-561, 2000.
#48-#55 based on Premilat & Albiser, ``A new D-DNA form of poly(dA-dT) .
poly(dA-dT): an A-DNA type structure with reversed Hoogsteen Pairing''.
Eur. Biophys. J., 30, pp. 404-410, 2001 (and several other publications).
Recently, I heard from Thomas Holder, the PyMOL Principal Developer (Schrödinger, Inc.), that he had written a wrapper to the 3DNA fiber command. This PyMOL wrapper is implemented as part of his versatile PSICO library (see the PyMOL Wiki page Psico for details), and exposes the 55 fiber models based on Arnott and other’s work to the wide PyMOL user community. Moreover, the wrapper can be accessed directly from PyMOL (without installing PSICO), as shown below with an example:
PyMOL> run https://raw.githubusercontent.com/speleo3/pymol-psico/master/psico/creating.py
PyMOL> fiber CTAGCG
The resulting fiber model is the default B-form DNA of calf thymus, with twist of 36.0° and rise of 3.375 Å (see figure below). Note that cases in base sequence do not matter, so fiber ctagcg or fiber CTAgcg will give the same result.

Running PyMOL>help fiber gives the following detailed usages info, which should be sufficient to get one started with this fiber tool in PyMOL.
PyMOL> help fiber
DESCRIPTION
Run X3DNA's "fiber" tool.
For the list of structure identification numbers, see for example:
http://xiang-jun.blogspot.com/2009/10/fiber-models-in-3dna.html
USAGE
fiber seq [, num [, name [, rna [, single ]]]]
ARGUMENTS
seq = str: single letter code sequence or number of repeats for
repeat models.
num = int: structure identification number {default: 4}
name = str: name of object to create {default: random unused name}
rna = 0/1: 0=DNA, 1=RNA {default: 0}
single = 0/1: 0=double stranded, 1=single stranded {default: 0}
EXAMPLES
# environment (this could go into ~/.pymolrc or ~/.bashrc)
os.environ["X3DNA"] = "/opt/x3dna-v2.3"
# B or A DNA from sequence
fiber CTAGCG
fiber CTAGCG, 1, ADNA
# double or single stranded RNA from sequence
fiber AAAGGU, name=dsRNA, rna=1
fiber AAAGGU, name=ssRNA, rna=1, single=1
# poly-GC Z-DNA repeat model with 10 repeats
fiber 10, 15
Thanks to Thomas, for making another connection between PyMOL and 3DNA/DSSR. The other one is the DSSR-plugin for PyMOL to create “block” shaped cartoons for nucleic acid bases and base pairs.
See also 3DNA fiber models
As of release v2.3-2016sept06, the C source code of the 3DNA software package is available. The code can be found in the $X3DNA/src folder of the distributed tarballs for Linux, Mac OS X, and Windows. Since 3DNA is written in pure ANSI C, it can be compiled without changes on any platform with a modern C compiler.
The original codebase of 3DNA was written around year 2000. Up until v2.3, the infrastructure of 3DNA has remained stable for 16 years. During the time, 3DNA has been widely adopted in other bioinformatics pipelines and cited over 1,500 times. Over the years, I’ve received quite a few requests for 3DNA source code. However, due to complications of various factors (including software licensing), 3DNA had only been distributed in executable forms for the crucial C programs. Now, the C code of 3DNA is finally open source!
As before, users need to register on the 3DNA Forum to download the software. The download page also includes x3dna-v2.0.tar.gz that accompanied the 2008 Nature Protocols paper, and x3dna-v1.5.tar.gz that corresponded to the 2003 Nucleic Acids Research paper. Other than minor revisions to pass strict gcc compiler options, the v1.5 and v2.0 codebases are kept as they were. 3DNA is backward-compatible as far as the key base-pair parameters are concerned. Moreover, between v1.5 and v2.0, the command-line interface stays the same. The two previous versions are released for historical reasons. For example, one may notice some obvious “similarities” between 3DNA v1.5 and RNAView.
The development of DSSR and SNAP will push 3DNA into a brand new version (v3), which contains significant changes in functionality and interface, and is no longer compatible with previous versions. I intend to keep 3DNA v2.3 in a ‘maintenance’ mode: no new features are planed, but bug reports and user questions will be promptly addressed on the 3DNA Forum, as always. Making 3DNA open source should help further prompt its adoptions, and adaptations in structural bioinformatics of nucleic acids.
There are numerous types of software licenses, but none of them seems to be a good fit for my purpose. As a result, I’ve come up with a permissive “citation-ware” license with contents as below:
3DNA is a suite of software programs for the analysis,
rebuilding and visualization of 3-Dimensional Nucleic Acid
structures. Permission to use, copy, modify, and distribute
this suite for any purpose, with or without fee, is hereby
granted, and subject to the following conditions:
At least one of the 3DNA papers must be cited, including the
following two primary ones:
1. Lu, X. J., & Olson, W. K. (2003). "3DNA: a software
package for the analysis, rebuilding and visualization
of three‐dimensional nucleic acid structures." Nucleic
Acids Research, 31(17), 5108-5121.
2. Lu, X. J., & Olson, W. K. (2008). "3DNA: a versatile,
integrated software system for the analysis,
rebuilding and visualization of three-dimensional
nucleic-acid structures." Nature Protocols, 3(7),
1213-1227.
THE 3DNA SOFTWARE IS PROVIDED "AS IS", WITHOUT EXPRESSED OR
IMPLIED WARRANTY OF ANY KIND.
Any 3DNA-related questions, comments, and suggestions are
welcome and should be directed to the open 3DNA Forum
(http://forum.x3dna.org/).
Upon user requests, I’ve recently introduced the --block-color option to DSSR, available as of v1.5.2-2016apr02. As its name implies, the --block-color option facilitate user customization of PyMOL rendered colors of the base rectangular blocks or their edges (e.g., the minor-groove) directly from the command-line. A simple example goes like this: --block-color='A blue; T red', which makes A colored blue and T colored red. As detailed below, the new option is very flexible with regard to the specification of colors, bases, or some edges to highlight. Before that, a little background is in order.
Background info
The DSSR cartoon-block representation follows the color convention of the original 3DNA blocview script, where A is red; C is yellow; G is green; T is blue; and U is cyan. If I remember correctly, the blocview coloring was based on the scheme adopted by the Nucleic Acid Database (NDB). To allow for some flexibility, 3DNA includes a config file named $X3DNA/config/raster3d.par where users can change the RGB values of the corresponding bases. However, I do not know if any user has ever bothered to play around with the configuration file for customized base colors.
Over the years, blocview-generated images have become popular, due to its simplicity, and (maybe more importantly) its endorsement by the NDB and PDB for nucleic acid structures. Via NDB, the blocview-generated images have also been used in RNA FRABASE 2.0 and RNA Structure Atlas. Nevertheless, the blocview script has several dependencies: MolScript for protein or DNA/RNA backbone ribbons, render from Raster3D for rendering, and ImageMagick for image processing. Moreover, the blocview script used by NDB/PDB is (likely to be) based on 3DNA v1.5, the last version before I left Rutgers in 2002.
Over the years, 3DNA has been continuously refined, with significant changes introduced in v2.0 around 2008 to accompany the Nature Protocols paper. Currently at v2.3, the codebase for 3DNA version 2 is in maintenance mode: the software will still be supported with identified bugs fixed, but no more new feature is planned. 3DNA version 3, as represented by DSSR and SNAP, is the way to go.
DSSR has no third-party dependencies
While creating DSSR, I set it as one of the design goals to make the program fully self-contained, without any third-party dependencies. Connections to other tools are clearly delineated via text files. If anything goes wrong, one can easily identify where the problem is. Experience over the past few years has unambiguously proved the effectiveness of this zero-dependency approach. Other than being directly distributed with an operating system, DSSR is the easiest to get up and running. Moreover, DSSR can be easily integrated into other pipelines, including Jmol and PyMOL, among many other bioinformatics tools.
For the cartoon-block representation, DSSR produces .r3d files that can be loaded into PyMOL, mixed and matched with other visualization styles PyMOL has to offer. No more direct dependencies on MolScript, Raster3D, and ImageMagick as is the case for blocview. It is also worth mentioning that DSSR does not need PyMOL to run. DSSR and PyMOL are connected via .r3d files, a process which can be streamlined with the Dssr_block PyMOL plugin.
DSSR releases before v1.5.2-2016apr02 have the color coding of base blocks fixed within the source code, following the default style of blocview. Over the past few months, I’ve received at least two explicit requests on customizing the default colors of DSSR-generated base blocks. The --block-color option has been introduced for this purpose.
Details of the --block-color option
The general format of the option is as follows:
--block-color='id color [; id2 color2 ...]'
id can be A, C, G, T, U, or the degenerated IUPAC code, including R, Y, N etc. See UPAC nucleotide code for details.
id can also be minor, major, upper, bottom, wc-edge to specify one of the six faces of a 3D rectangular block. See Fig.1D of the DSSR paper for details.
id can further be GC, AT, GU, pair, and variants thereof, to specify the colors of the corresponding long base-pair rectangular blocks.
color can be a common name (144 total), as specified in the RGB Color website. For example, red, magenta, light gray etc.
color can also be a single number in the range [0, 1] or [0, 255] to specify a shade of gray. DSSR repeat the number twice to get the RGB triple consisting of the same number.
color can further be a set of three _space_-delimited numbers to specify the RGB triple. Again, the number can be in [0, 1] or [0, 255]. Moreover, the three numbers can be put in square brackets. For example --block-color='A 0 1 1' and --block-color='A [0 1 1]' specify adenine to be colored with RGB triple [0 1 1] (aqua/cyan, corresponding to --block-color='A cyan').
- More than one identity (bases) can be specified, separated by
; (,, :, or | also works). Note: within the PyMOL dssr_block plugin, only | or : can be used as a separator: comma (,) or semicolon (;) cannot be used as a separator within a PyMOL command argument (thanks to Thomas Holder for drawing this point to my attention).
- Case does not matter when specifying
id or color. So either ‘A’ or ‘a’, and ‘blue’ or ‘Blue’ or ‘BLUE’ can be used to make adenine blue: --block-color='a blue'.
Some example usages
While the above description may appears to be quite complicated, the actual usage of the --block-color option is very straightforward. As always, the cases are best made with concrete examples, as shown below using the classic Dickerson B-DNA dodecamer 355d.
# all bases in blue
x3dna-dssr -i=355d.pdb --cartoon-block=orient --block-color='N blue' -o=355d-all-blue.pml
#
# all WC-pairs in red, with the minor-groove edge in 'dim gary'
x3dna-dssr -i=355d.pdb --cartoon-block=orient --block-color='wc-pair red; minor dim gray' -o=355d-pair-minor.pml
#
# thymine (T) in purple, and the upper (+z) face in white
# see Figure below, which shows the two bases in WC-pairs are anti-parallel
x3dna-dssr -i=355d.pdb --cartoon-block=orient --block-color='T purple; upper 1' -o=355d-T-upper.pml
 faces white")
{kind=link}
{kind=link}