Recently, I came across the paper by Mitra et al. (2025) titled "RNAproDB: A Webserver and Interactive Database for Analyzing Protein-RNA Interactions." I am glad to notice that DSSR (Lu et al. 2015) has been cited extensively in this work, as follows:
As part of the processing pipeline, multiple software is run including DSSR^12^ (base-pairing geometries, protein–RNA hydrogen bonds, and RNA secondary structure), HBPLUS^17^ (hydrogen bonds involving water molecules), ... Leontis-Westhof^27^ base pair annotations (as computed by DSSR^12^) ... The structural elements (stems, loops, hairpins, junctions, etc.) are detected using DSSR^12^ and mapped to the partial projection layout (via averaging corresponding residue coordinates)... We explored the relative abundance of different standard nucleotides (A, C, G, and U) in helical vs. non-helical regions (as computed by DSSR^12^)...We quantified the propensity of base-pairing (as detected by DSSR^12^) between different RNA bases (Fig. 3D).
This is an impressive contribution on the characterization of protein-RNA interactions. Reading carefully through the paper and its supplemental PDF, I was intrigued by the following note on a water-mediated U-U base pair missed by DSSR.
Another important aspect to discuss is RNA–RNA water-mediated interactions^33,34^. ... One such example is the CUG repeat structure from PDB ID 7Y2B^35^ (Fig. S5A). The U/U mismatches in this structure are often unable to form direct hydrogen bonds (specifically, the central U/U mismatch forms no direct hydrogen bond). Therefore, DSSR^12^ does not classify it as a base pair. However, two water molecules form water-mediated hydrogen bonds between the two U bases. ...
While DSSR internally already takes consideration of water-mediated H-bonds in the detection of base pairs, it still requires: (1) at least one direct H-bond between two base atoms or a base atom to backbone, and (2) a co-planar geometry between the two bases. The water-mediated U7-U7 pair in PDB entry 7Y2B does not fulfill condition (1): the minimal distance between the two U bases is 5 Å, which is far larger than a typical H-bonding distance. Therefore, DSSR did not classify it as a base pair.
Prompted by the observation of Mitra et al. (2025), I have added a new option (--pair-water) in the DSSR v2.5.1-2025mar19 release to allow for water-mediated base pairs to be detected. Using PDB entry 7Y2B as an example, the DSSR command and related base-pairs output are shown below.
# x3dna-dssr -i=7Y2B.pdb1 --symm --pair-water
List of 13 base pairs
nt1 nt2 bp name Saenger LW DSSR
1 1:S.U1 2:S.A13 U-A WC 20-XX cWW cW-W
2 1:S.U2 2:S.A12 U-A WC 20-XX cWW cW-W
3 1:S.C3 2:S.G11 C-G WC 19-XIX cWW cW-W
4 1:S.U4 2:S.U10 U-U -- -- cWW cW-W
5 1:S.G5 2:S.C9 G-C WC 19-XIX cWW cW-W
6 1:S.C6 2:S.G8 C-G WC 19-XIX cWW cW-W
7 1:S.U7 2:S.U7 U-U Water -- cWW cW-W
8 1:S.G8 2:S.C6 G-C WC 19-XIX cWW cW-W
9 1:S.C9 2:S.G5 C-G WC 19-XIX cWW cW-W
10 1:S.U10 2:S.U4 U-U -- -- cWW cW-W
11 1:S.G11 2:S.C3 G-C WC 19-XIX cWW cW-W
12 1:S.A12 2:S.U2 A-U WC 20-XX cWW cW-W
13 1:S.A13 2:S.U1 A-U WC 20-XX cWW cW-W
Base pair #7 is water-mediated, as shown in the molecular image below. Note that .pdb1 means biological unit 1, and the option --symm reads the two symmetry-related structures in the MODEL/ENDMDL delineated ensemble as a single structure. See the DSSR User Manual for more details.

References
-
Lu, Xiang-Jun, Harmen J. Bussemaker, and Wilma K. Olson. 2015. “DSSR: An Integrated Software Tool for Dissecting the Spatial Structure of RNA.” Nucleic Acids Research, July, gkv716. https://doi.org/10.1093/nar/gkv716.
- Mitra, Raktim, Ari S. Cohen, Wei Yu Tang, Hirad Hosseini, Yongchan Hong, Helen M. Berman, and Remo Rohs. 2025. “RNAproDB: A Webserver and Interactive Database for Analyzing Protein-RNA Interactions.” Journal of Molecular Biology, February, 169012. https://doi.org/10.1016/j.jmb.2025.169012.

DSSR produces RNA secondary structures in connect table (.ct) format. According to "RNAstructure Command Line Help: File Formats" (with slight editing):
CT File Format
A CT (Connectivity Table) file contains secondary structure information for a sequence. These files are saved with a CT extension. When entering a structure to calculate the free energy, the following format must be followed.
- Start of first line: number of bases in the sequence
- End of first line: title of the structure
- Each of the following lines provides information about a given base in the sequence. Each base has its own line, with these elements in order:
- Base number: index n
- Base (A, C, G, T, U, X)
- Index n-1
- Index n+1
- Number of the base to which n is paired. No pairing is indicated by 0 (zero).
- Natural numbering. RNAstructure ignores the actual value given in natural numbering, so it is easiest to repeat n here.
Using PDB entry 1msy as an example (see Figure 1 below):
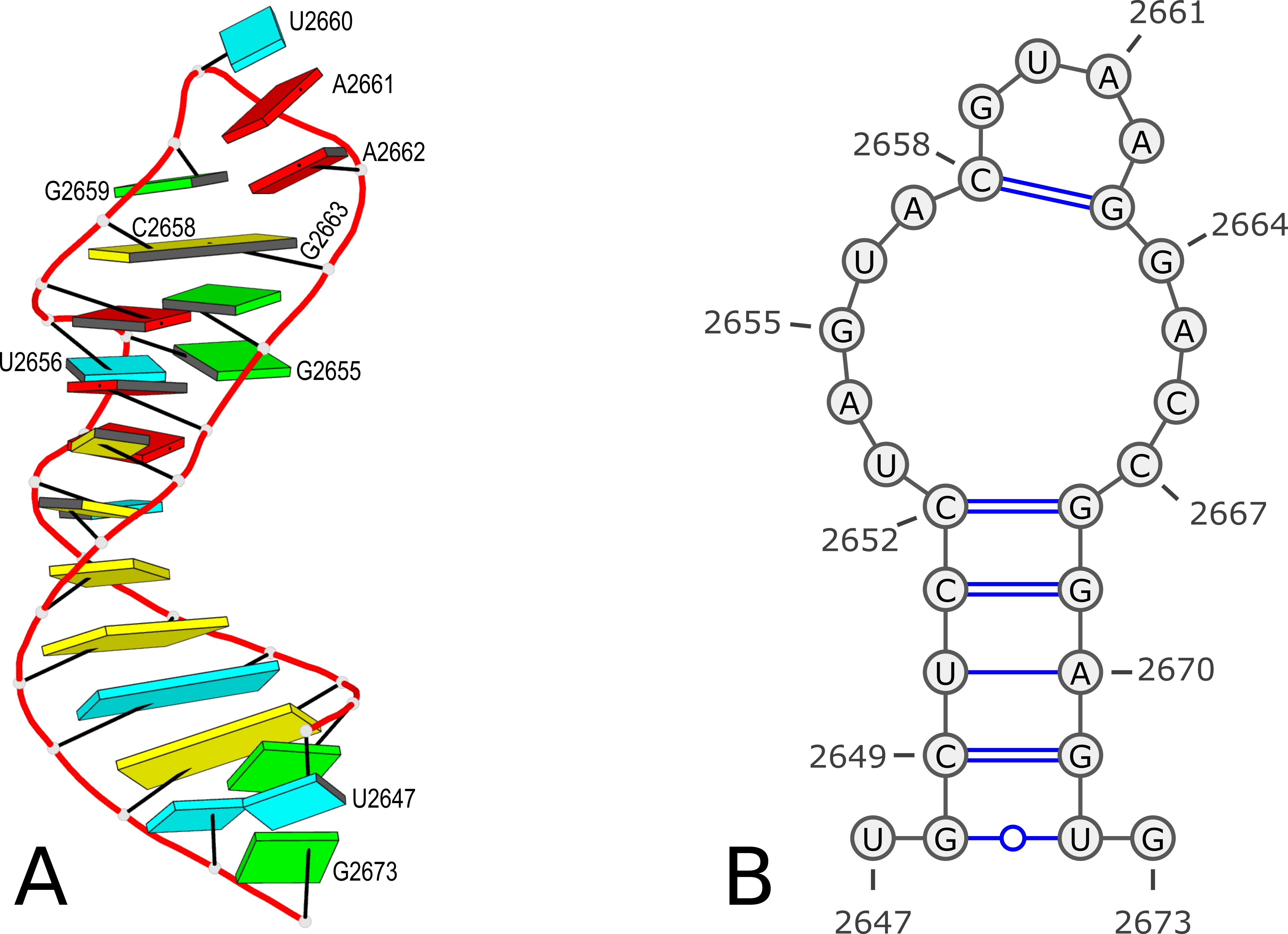
Figure 1. 3D and 2D structures of PDB entry 1msy. (A) 3D schematic auto-created via the DSSR-PyMOL integration. The labeled residues follow PDB coordinates. (B) 2D diagram rendered with VARNA using DSSR-derived 2D structural information in the .ct format. This figure was annotated using Inkscape.
With commands:
x3dna-dssr -i=1msy.pdb
cp dssr-2ndstrs.ct 1msy-dssr-default.ct
The file 1msy-dssr-default.ct has the following contents:
27 ENERGY = 0.0 [1msy] -- secondary structure derived by DSSR
1 U 0 2 0 2647
2 G 1 3 26 2648
3 C 2 4 25 2649
4 U 3 5 24 2650
5 C 4 6 23 2651
6 C 5 7 22 2652
7 U 6 8 0 2653
8 A 7 9 0 2654
9 G 8 10 0 2655
10 U 9 11 0 2656
11 A 10 12 0 2657
12 C 11 13 17 2658
13 G 12 14 0 2659
14 U 13 15 0 2660
15 A 14 16 0 2661
16 A 15 17 0 2662
17 G 16 18 12 2663
18 G 17 19 0 2664
19 A 18 20 0 2665
20 C 19 21 0 2666
21 C 20 22 0 2667
22 G 21 23 6 2668
23 G 22 24 5 2669
24 A 23 25 4 2670
25 G 24 26 3 2671
26 U 25 27 2 2672
27 G 26 0 0 2673
Here the first line contains 27 (as the number of bases) and ENERGY = 0.0 [1msy] -- secondary structure derived by DSSR (as the title). While RNAstructure ignores the actual values given in natural numbering, DSSR outputs the residue numbers of the nucleotides (e.g. U2467 and G2673) in the PDB file.
With the DSSR option --structure-title (or --str-title, actually via regex "^-?-?str(ucture)?[-_]?title"), users can set the title for the derived .ct file, as shown below:
x3dna-dssr -I=1msy.pdb --structure-title='CT file derived from DSSR'
cp dssr-2ndstrs.ct 1msy-dssr-title.ct
27 CT file derived from DSSR
1 U 0 2 0 2647
2 G 1 3 26 2648
......
26 U 25 27 2 2672
27 G 26 0 0 2673
One can also remove the title, by using an empty string "" (i.e., --str-title="") or simply --str-title (or --str-title=).
x3dna-dssr -I=1msy.pdb --structure-title=""
cp dssr-2ndstrs.ct 1msy-dssr-notitle.ct
27
1 U 0 2 0 2647
2 G 1 3 26 2648
......
With the --more option, DSSR also outputs additional info that can be used to easily identify a nucleotide and its pairing partner.
x3dna-dssr -I=1msy.pdb --more --structure-title="1msy with extra info"
cp dssr-2ndstrs.ct 1msy-dssr-extra.ct
27 1msy with extra info
1 U 0 2 0 2647 # name=A.U2647
2 G 1 3 26 2648 # name=A.G2648, pairedNt=A.U2672
3 C 2 4 25 2649 # name=A.C2649, pairedNt=A.G2671
......
Note that unlike for the .bpseq format with extra info which cannot be fed directly into VARNA, the extra info for the .ct format causes no troubles for VARNA to visualize the 2d structure.
The --structure-title option is another small feature implemented in DSSR. It is currently not documented in the DSSR User Manual since this feature is unlikely of general interest.
DSSR commands used, and the output .ct files:
x3dna-dssr -i=1msy.pdb
cp dssr-2ndstrs.ct 1msy-dssr-default.ct
x3dna-dssr -I=1msy.pdb --structure-title='CT file derived from DSSR'
cp dssr-2ndstrs.ct 1msy-dssr-title.ct
x3dna-dssr -I=1msy.pdb --structure-title=""
cp dssr-2ndstrs.ct 1msy-dssr-notitle.ct
x3dna-dssr -I=1msy.pdb --more --structure-title="1msy with extra info"
cp dssr-2ndstrs.ct 1msy-dssr-extra.ct
By default, DSSR produces RNA secondary structures in three commonly used file formats––ViennaRNA package dbn, Mfold connect table (.ct), and CRW bpseq––that can be fed directly into visualization tools such as VARNA. In this blog post, I want to dig deeper into the bpseq format, and show the variations available from DSSR.
According to "RNA STRAND v2.0 - The RNA secondary STRucture and statistical ANalysis Database" (with slight editing):
BPSEQ format:The file name should end with the suffix ".bpseq", as in "mystr.bpseq". The bpseq format is a simple text format in which there is one line per base in the molecule, listing the position of the base (leftmost position is 1), the base name (A,C,G,U, or other alphabetical characters), and the position number of the base to which it is paired, with a 0 denoting that the base is unpaired. For more information, see the Comparative RNA Web Site. An example is as follows:
1 G 8
2 G 7
3 C 0
4 A 0
5 U 0
6 U 0
7 C 2
8 C 1
For complexes with more than one molecule, the molecules are listed in sequence, with the base pairs numbers of each successive molecule following in order from the previous molecule.
The bases in bpseq format are identified by position numbers starting from 1 for the leftmost position. That is the convention DSSR follows by default in its .bpseq output. For example, for the PDB entry 1msy, which contains 27 nucleotides, the command x3dna-dssr -i=1msy.pdb will generate a file named dssr-2ndstrs.bpseq with the following contents (abbreviated):
1 U 0
2 G 26
3 C 25
......
25 G 3
26 U 2
27 G 0
However, according to PDB atomic coordinates, the nucleotides are numbered from U2647 (#1) to G2673 (#27) as shown in the Figure 1 below:
Figure 1. 3D and 2D structures of PDB entry 1msy. (A) 3D schematic auto-created via the DSSR-PyMOL integration. The labeled residues follow PDB coordinates. (B) 2D diagram rendered with VARNA using DSSR-derived 2D structural information in the .ct format. This figure was annotated using Inkscape.
It makes sense that the labelling of bases in the 2D bpseq format follows those from the 3D atomic coordinates in the PDB. Thus instead of starting from position 1 as shown above, the bpseq file would start with 2647. That's exactly what the DSSR --bpseq option is created for. Thus, with the command x3dna-dssr -i=1msy.pdb --bpseq, the output file dssr-2ndstrs.bpseq now has the following contents (abbreviated):
2647 U 0
2648 G 2672
2649 C 2671
......
2671 G 2649
2672 U 2648
2673 G 0
This .bpseq file can be read by VARNA (tested with VARNAv3-93.jar) to generate a 2D image as shown in Figure 1(B) above.
Moreover, with the command x3dna-dssr -i=1msy.pdb --bpseq=extra, the output file dssr-2ndstrs.bpseq now contains additional info to easily identify a nucleotide and its pairing partner:
2647 U 0 # name=A.U2647
2648 G 2672 # name=A.G2648, pairedNt=A.U2672
2649 C 2671 # name=A.C2649, pairedNt=A.G2671
......
2671 G 2649 # name=A.G2671, pairedNt=A.C2649
2672 U 2648 # name=A.U2672, pairedNt=A.G2648
2673 G 0 # name=A.G2673
It should be noted that this .bpseq output file is no longer compliant to the standard, and can not be fed into VARNA for visualization.
The --bpseq option has been added upon users' request. The --bpseq=extra variation was implemented recently to ensure that the --bpseq option by itself produce a valid .bpseq file without extra info (e.g., enabled with the global --more option). Now the extra info for .bpseq output is enabled only by setting --bpseq=extra explicitly.
This --bpseq option and its evolution is a good example of how DSSR responds to community requests. I'm here to listen and I'm always willing to improve DSSR that better fit users' needs. If you make use of DSSR in your pipeline and need some adaptions, please do not hesitate to contact me. I may consider adding a new option or revising the code otherwise that would streamline the integration of DSSR into your project.
DSSR commands used, and the output .bpseq files:
x3dna-dssr -i=1msy.pdb
cp dssr-2ndstrs.bpseq 1msy-dssr-default.bpseq
x3dna-dssr -i=1msy.pdb --bpseq
cp dssr-2ndstrs.bpseq 1msy-dssr-bpseq.bpseq
x3dna-dssr -i=1msy.pdb --bpseq=extra
cp dssr-2ndstrs.bpseq 1msy-dssr-bpseq-extra.bpseq
As I am writing this blogpost on June 26, 2020, the registrations on the 3DNA Forum has reached 5,054. The numbers were 3,000 on October 15, 2016, 2,000 on on February 3, 2015, and 1,000 on February 27, 2013 respectively. For year 2020, the monthly registrations are 36 (January), 35 (February), 54 (March), 84 (April), 69 (May). As of June 26, the number is 56, which will more than likely pass 60 by the end of this month. The Covid-19 pandemic does not seem to having a negative effect on the registrations.
The over 5,000 registrations are from users all over the world. The 3DNA Forum remains spam free, and all questions are promptly answered. It is functioning well; certainly better than I originally imagined.
Overall, the Forum serves as a virtual platform for me to interact effectively with the ever-increasing user community. I greatly enjoy answering questions, fixing bugs, and making 3DNA/DSSR/SNAP better tools for real-world applications.
In late March, I was approached by Mike May. He was then writing an article for Biocompare about DNA-protein interactions and asked me to answer a few questions on “What features of 3DNA be used in studying DNA-protein interactions?” and “Please provide 1-2 examples.” Initially, I was a bit surprised by the contact. Thus, I visited his online profile and Amazon Author Page. I also read a couple of his previous publications. Impressed by his track records, I answered his requests and our following communications were as smooth and professional as I could have ever imagined.
The paper The Best Ways to Study DNA and Protein Interactions has now been published, and is freely accessible. It includes the following content:
3DNA creator and maintainer Xiang-Jun Lu mentioned a couple of ways that the software has been used. For example, he noted that “3DNA can analyze all DNA-protein complexes in the Protein Data Bank—PDB—in an automatic, consistent, and robust manner,” and other bioinformatic resources have adopted this feature of 3DNA. He added that scientists have used 3DNA to “understand the structural basis on how transcription factors recognize methylated DNA.” Moreover, 3DNA is continuously developed. A new feature of 3DNA is the automatic identification and comprehensive characterization of G-quadruplexes, a noncanonical DNA structure formed from guanine-rich base sequences.
The bioinformatics resource I used as an example is the paper DNAproDB: an expanded database and web-based tool for structural analysis of DNA–protein complexes by the Rohs lab. The phrase “to understand the structural basis on how transcription factors recognize methylated DNA” refers to the article Toward a mechanistic understanding of DNA methylation readout by transcription factors by the Bussemaker lab. Both works employed DSSR and SNAP, two sophisticated programs I created and maintained over the past ten years, and they have largely obsoleted the original 3DNA suite of programs.
The image I provided is a DSSR-PyMOL schematic based on PDB entry 6LDM. The 6LMD picture features a G-quadruplex, for which DSSR comes with an unmatched set of features (including automatic identification and comprehensive annotations). See the http://g4.x3dna.org/ page for survey results, curated using DSSR, of all G-quadruplexes from the PDB.
This image of a protein-DNA complex (PDB entry 6LDM) shows the protein (purple), the DNA G-quadruplex (green) and thymine (blue). The image was created using the 3DNA-DSSR program and PyMOL. Image courtesy of Xiang-Jun Lu.

A couple of months ago, I came across the homepage of the newly-established G4 Society on G-quadruplexes (G4s). I checked the “Online tools” section and found a few links to G4 databases and sequence-based predication programs (e.g., G4Hunter). No tools, however, were listed for G4 identification and characterization from 3D atomic coordinates as those deposited in the Protein Data Bank (PDB). So I filled out the contact form and provided a brief description of 3DNA-DSSR, including a link to the website of G4s auto-curated with DSSR from the PDB.
I’ve recently visited the G4-society website again. I am pleased to see that 3DNA-DSSR is now listed under Online tools as a “program for detections/annotations of G4 from atomic coordinates in PDB or PDBx/mmCIF format”. The G4 module of 3DNA-DSSR has been created to streamline the identification and annotation of 3D structures of G4s. The collection of G4s in the PDB, available at G4.x3dna.org, is updated weekly. It represents a unique resource for the G4 community. Hopefully, its value will be more widely appreciated thanks to the link from the G4-society website.
At the G4-society homepage, I noticed the following two items in the “News” section (on December 13, 2019):
The Quadruplex Meeting Report
Meeting report: Seventh International Meeting on Quadruplex Nucleic Acids (Changchun, P.R. China, September 6e9, 2019) written by Jean-Louis Mergny. Reading through the report, I noticed the following:
Jonathan B. Chaires (U. Louisville, KY, USA) provided an overview and historical perspective of the quadruplex field in his inaugural lecture. As of August 2019, the quadruplex field gathers 8467 articles and 253,174 citations in the Science Citation Index. Over 200 G4 structures are available in the PDB.
I did not know how the survey of G4s in the PDB was performed. Based on my data, the PDB-G4 structures was already over 300 as of August 2019. As of December 11, 2019, the number of G4 structures in the PDB is 329. Importantly, the PDB-G4 website compiled using 3DNA-DSSR contains not only citation information but also detailed annotations and schematic images not available elsewhere. Here are a few recent examples:
- PDB id: 6ge1 — “Unraveling the structural basis for the exceptional stability of RNA G-quadruplexes capped by a uridine tetrad at the 3’ terminus.” by Andralojc et al. in RNA (2019).
- PDB id: 6gh0 — “Two-quartet kit* G-quadruplex is formed via double-stranded pre-folded structure.” by Kotar et al. in Nucleic Acids Res. (2019).
- PDB id: 6e8u — “Structure and functional reselection of the Mango-III fluorogenic RNA aptamer.” by Trachman et al. in Nat. Chem. Biol. (2019).
- PDB id: 6ac7 —“Structure of a (3+1) hybrid G-quadruplex in the PARP1 promoter.” by Sengar et al. in Nucleic Acids Res. (2019).
The Important Paper
A guide to computational methods for G-quadruplex prediction by Emilia Puig Lombardi and Arturo Londoňo-Vallejo in Nucleic Acids Res. (2019), which presents an updated overview of G4 prediction algorithms. I am impressed by the large number of sequence-based G4 prediction software tools, including the most recent G4-iM Grinder. Nevertheless, as noted by the authors in the concluding remarks, “All computational G-quadruplex prediction approaches have their drawbacks and limitations despite the recent advances in the field and the introduction of validation steps based on experimental data.”
The G4 module in 3DNA-DSSR belongs to a completely different category of software tool. It does not ‘predict’ G4 propensity/stability from a base sequence, but identify and annotate G4s in a 3D atomic coordinate file. It complements sequence-based predicting tools by gaining insights into the 3D G4 structures and refining folding rules to improve performance of prediction tools. Based on my knowledge, the 3D G4 structures contains features that are not captured by any of the sequence-based prediction tools.
While reading the review article, I found Fig. 1 informative (see below). The right side of Fig. 1A shows a “cartoon representation of the Oxytricha telomeric DNA G4 crystal structure (PDB accession 1JPQ (112))” using PyMOL. In comparison, the cartoon-block image auto-generated via 3DNA-DSSR and PyMOL for PDB id: 1jpq is shown at the bottom. The DSSR-PyMOL version is obviously different, presumably simpler and more informative, from that illustrated in Fig. 1A.


Recently I read the article Topology-based classification of tetrads and quadruplex structures in Bioinformatics by Popenda et al. In this work, the authors proposed an ONZ classification scheme of G-tetrads in intramolecular G-quadruplexes (G4) as shown below (Fig. 2 in the publication):

I am glad to find that DSSR has been used as a component in their computational tool ElTetrado to automatically identify and classify tetrads and quadruplexes.
Structures from both sets were analysed using self-implemented programs along with DSSR software from the 3DNA suite (Lu et al. (2015)). From DSSR, we acquired the information about base pairs and stacking.
I like the ONZ classification scheme: it is simple in concept yet provides a new perspective for the topologies of G-tetrads in intramolecular G4 structures. So I implemented the idea in DSSR v1.9.8-2019oct16, with this feature available via the --g4-onz option. Note that ElTetrado, according to the authors, is applicable to ONZ classifications of general types of tetrads and quadruplexes. The DSSR implementation of ONZ classifications, on the other hand, is strictly limited to G-tetrads in intramolecular G4 structures.
The DSSR ONZ classification results match the ones reported in Figs. 1, 5, and 6 of the Popenda et al. paper. For example, for PDB entry 6H1K (Fig. 6), the relevant results with the --g4-onz option and without it are listed below:
# x3dna-dssr -i=6h1k.pdb --g4-onz
List of 3 G-tetrads
1 glyco-bond=s--- groove=w--n planarity=0.149 type=planar Z- nts=4 GGGG A.DG1,A.DG20,A.DG16,A.DG27
2 glyco-bond=-sss groove=w--n planarity=0.136 type=planar Z+ nts=4 GGGG A.DG2,A.DG19,A.DG15,A.DG26
3 glyco-bond=--s- groove=-wn- planarity=0.307 type=other O+ nts=4 GGGG A.DG17,A.DG21,A.DG25,A.DG28
# ---------------------------------------
# x3dna-dssr -i=6h1k.pdb
# without option --g4-onz
List of 3 G-tetrads
1 glyco-bond=s--- groove=w--n planarity=0.149 type=planar nts=4 GGGG A.DG1,A.DG20,A.DG16,A.DG27
2 glyco-bond=-sss groove=w--n planarity=0.136 type=planar nts=4 GGGG A.DG2,A.DG19,A.DG15,A.DG26
3 glyco-bond=--s- groove=-wn- planarity=0.307 type=other nts=4 GGGG A.DG17,A.DG21,A.DG25,A.DG28
With the --json option, the ONZ classification results are always available. An example is shown below for PDB entry 6H1K (Fig. 6):
# x3dna-dssr -i=6h1k.pdb --json | jq -c '.G4tetrads[] | [.nts_long, .topo_class]'
["A.DG1,A.DG20,A.DG16,A.DG27","Z-"]
["A.DG2,A.DG19,A.DG15,A.DG26","Z+"]
["A.DG17,A.DG21,A.DG25,A.DG28","O+"]
I recently read a short communication by Pavel Afonine, titled phenix.hbond: a new tool for annotation hydrogen bonds in the July 2019 issue of the Computational Crystallography Newsletter (CCN). It appears that every bioinformatics tool (e.g., PyMOL or Jmol) has its own implementation of an algorithm on calculating H-bonds, one of the fundamental stabilizing forces of proteins and DNA/RNA structures. So does 3DNA/DSSR, as noted in my 2014-04-11 blogpost Get hydrogen bonds with DSSR.
Both DSSR and SNAP have the --get-hbond option, and they use the same underlying algorithm. However, the default output from the two programs differs: DSSR reports the H-bonds within nucleic acids, and SNAP covers only those at the DNA/RNA-protein interface. Using the PDB entry 1oct as an example, Running DSSR on it with the --get-hbond option gives 33 H-bonds in the DNA duplex, while SNAP reports 38 H-bonds at the DNA-protein interface. By design, the default output caters for the most-common use case of each program.
Under the scene, however, there exist variations in the seemingly simple --get-hbond option. One can attach text ‘nucleic’ (or ‘nuc’, ‘nt’), as in --get-hbond-nucleic, to output H-bonds within nucleic acids. Similarly, --get-hbond-protein (or ‘amino’, ‘aa’) would output H-bonds within proteins. Not surprisingly, the --get-hbond-nt-aa option would list H-bonds in nucleic acids and proteins, including those at their interface. These variations apply to both DSSR and SNAP, even though some are redundant with the default.
Notably, in combination with --json, the --get-hbond option by default would output all H-bonds, as if --get-hbond-nt-aa has been set. For PDB entry 1oct, DSSR or SNAP would report 208 H-bonds. Moreover, the JSON output has a residue_pair field for each identified H-bond, with values like "nt:nt", "nt:aa", or "aa:aa". Using 1oct as an example,
# x3dna-dssr -i=1oct.pdb --get-hbond --json | jq '.hbonds[0]'
{
"index": 1,
"atom1_serNum": 34,
"atom2_serNum": 608,
"donAcc_type": "standard",
"distance": 3.304,
"atom1_id": "O6@A.DG202",
"atom2_id": "N4@B.DC230",
"atom_pair": "O:N",
"residue_pair": "nt:nt"
}
# x3dna-dssr -i=1oct.pdb --get-hbond --json | jq '.hbonds[60]'
{
"index": 61,
"atom1_serNum": 462,
"atom2_serNum": 1187,
"donAcc_type": "standard",
"distance": 3.692,
"atom1_id": "O2@B.DT223",
"atom2_id": "NH2@C.ARG102",
"atom_pair": "O:N",
"residue_pair": "nt:aa"
}
# x3dna-dssr -i=1oct.pdb --get-hbond --json | jq '.hbonds[100]'
{
"index": 101,
"atom1_serNum": 791,
"atom2_serNum": 818,
"donAcc_type": "standard",
"distance": 2.871,
"atom1_id": "N@C.THR26",
"atom2_id": "OD2@C.ASP29",
"atom_pair": "N:O",
"residue_pair": "aa:aa"
}
In the above three cases, using SNAP instead of DSSR would give the same results.
Also, one can take advantage of the residue_pair value to filter H-bonds by type. For example, the following command would extract only H-bonds at the DNA-protein interface (38 occurrences, same as the number noted above):
x3dna-snap -i=1oct.pdb --get-hbond --json | jq '.hbonds[] | select(.residue_pair=="nt:aa")'
Back to the phenix.hbond tool, the author noted that:
Running phenix.hbond requires atomic model in PDB or mmCIF format with all hydrogen atoms added, as well as ligand restraint files if the model contains unknown to the library items.
While there is no particular reason why this should not work for all bio-macromolecules, currently phenix.hbond is only optimized and tested to work with proteins, which is the limitation that will be removed in future.
In contrast, the H-bond identification algorithm in DSSR/SNAP does not require hydrogen atoms. In fact, hydrogen atoms are simply ignored if they exist. As shown above, the H-bond method as implemented in DSSR/SNAP works for DNA, RNA, protein, or their complexes. This does not necessarily mean that the 3DNA way is superior to other similar tools. It just works well in my hand, and it may serve as a pragmatic choice for other users.
In the PDB, the ligand identifiers 5MC and 5CM all refer to 5-methylcytosine, but differ in the sugar moieties the base is attached to. Chemically, 5CM is 5-methyl-2’-deoxycytidine-5’-monophosphate as in DNA, and 5MC is 5-methylcytidine-5’-monophosphate. See the molecular images shown below.

The 5-methyl group is named C5A in 5CM and CM5 in 5MC, respectively, for non-obvious reasons other than conventions. For comparison, the methyl-group in thymine of DNA is named C7, as for example in PDB id 355d. It is worth noting that DSSR is able to handle all such variations in atom or residue names.