
Crystal structure of SARS-CoV-2 stem–loop 5 (SL5) (PDB id: 9E9Q; Jones CP, Ferré-D'Amaré AR. 2025. Crystallographic and cryoEM analyses reveal SARS-CoV-2 SL5 is a mobile T-shaped four-way junction with deep pockets. RNA 31: 949–960). The T-shaped four-way junction of the coronavirus SL5 structural element provides a starting point for examining the structures of larger RNA motifs and their interactions with other molecules. Image highlighting the four arms of the junction. The RNA backbone is depicted by a gray ribbon. The bases within the arms of the junction are colored respectively in blue, red, yellow, and cyan. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for Structural Bioinformatics of Nucleic Acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
As the developer of DSSR, I am thrilled to see its application in cutting-edge research across multiple disciplines. Below is a list of four recent publications that highlight how DSSR has been utilized, underscoring its versatility and significance in structural bioinformatics.
In the Geng et al. (2025) Nucleic Acids Research (NAR) paper, titled 'Revealing hidden protonated conformational states in RNA dynamic ensembles', DSSR is simply cited as follows:
All bp geometries, hydrogen-bond, backbone, stacking, and sugar dihedral angles were calculated using X3DNA-DSSR [77].
In the preprint by Gordan et al. (2025), titled 'High-throughput characterization of transcription factors that modulate UV damage formation and repair at single-nucleotide resolution', DSSR is cited as follows:
Step base stacking, base pair shift, base pair slide, interbase angle, pseudorotation angle, and sugar puckering classifications of nucleobases were computed using X3DNA-DSSR (v2.5.0)75. Base stacking was defined as the overlapping polygon area in Å2 when projecting the dipyrimidine base ring atoms (excluding exocyclic atoms) into the mean base pair plane76. The sugar ring pseudorotation phase angle of each pyrimidine was also calculated using X3DNA-DSSR as described by Altona, C. & Sundaralingam, M.77 Interbase angle was defined as sqrt(propeller2+buckle2) per the X3DNA-DSSR documentation.
Figure 6: TF Binding Induces Structural Distortion Favorable to UV Dimerization is highly informative, particularly panel (a), which illustrates the ensemble of structural parameters that predispose dipyrimidines to cyclobutane pyrimidine dimers (CPD) or 6-4 pyrimidine-pyrimidones (6-4 PP) formation. DSSR is designed as an integrated software tool, offering a comprehensive suite of structural parameters not found in any other single tool I am aware of. Despite this, the innovative use of DSSR by Gordan et al. exceeds my expectations and demonstrates its versatility.
In the preprint by Kubaney et al. (2025) from the Baker group, titled 'RNA sequence design and protein-DNA specificity prediction with NA-MPNN', DSSR is cited as follows:
On the pseudoknot subset, we evaluate additional structure‐ and reactivity‐based metrics. DSSR v2.3.241 is used to extract the ground‐truth secondary structure from the native crystal structures. For each designed sequence, RibonanzaNet predicts 2A3 reactivity profiles, from which we compute predicted OpenKnot scores (see https://github.com/eternagame/OpenKnotScore)31 using the predicted reactivity together with the DSSR ground truth.
In a recent NSMB paper from the Baker group, titled 'Computational design of sequence-specific DNA-binding proteins', 3DNA is cited as follows:
RIF docking of scaffolds onto DNA targets (DBP design step 1) Structures of B-DNA for each target (Supplementary Table 2) were generated by (1) using the DNA portion of PDB 1BC8 (ref. 60), PDB 1YO5 (ref. 61), PDB 1L3L (ref. 51) or PDB 2O4A (ref. 62) or (2) using the software X3DNA63, followed by a constrained Rosetta relax of the DNA structure.
Please note that 3DNA has been replaced by DSSR. The functionality for constructing B-DNA models, previously provided by 3DNA, is now directly available in DSSR via its fiber and rebuild modules.
In the preprint by Si et al. (2025), titled 'End-to-End Single-Stranded DNA Sequence Design with All-Atom Structure Reconstruction', DSSR is cited as follows:
Since ViennaRNA and NUPACK require secondary structures as input, we used DSSR35 to extract secondary structures from the corresponding ssDNA three-dimensional structures.
The above use cases are merely a sample of how DSSR is utilized in the scientific literature. It is reasonable to state that DSSR has emerged as a de facto standard tool within the field of nucleic acid structural bioinformatics. Overall, DSSR is a mature, robust, and efficient software product that is actively developed and maintained. I am committed to making DSSR synonymous with quality and value. Its unmatched functionality, usability, and support save users significant time and effort compared to alternative solutions.
DSSR is available free of charge for academic users. Additionally, it has been integrated into other high-profile bioinformatics resources, including NAKB, PDB-redo, and N•ESPript.
References
- Geng A, Roy R, Ganser L, Li L, Al-Hashimi HM. Revealing hidden protonated conformational states in RNA dynamic ensembles. Nucleic Acids Research. 2025;53:gkaf1366. https://doi.org/10.1093/nar/gkaf1366.
- Gordan R, Wasserman H, Chi B, Bohm K, Duan M, Sahay H, et al. High-throughput characterization of transcription factors that modulate UV damage formation and repair at single-nucleotide resolution. 2025. https://doi.org/10.21203/rs.3.rs-8197218/v1.
- Kubaney A, Favor A, McHugh L, Mitra R, Pecoraro R, Dauparas J, et al. RNA sequence design and protein–DNA specificity prediction with NA-MPNN. 2025. https://doi.org/10.1101/2025.10.03.679414.
- Glasscock CJ, Pecoraro RJ, McHugh R, Doyle LA, Chen W, Boivin O, et al. Computational design of sequence-specific DNA-binding proteins. Nat Struct Mol Biol. 2025;32:2252–61. https://doi.org/10.1038/s41594-025-01669-4.
- Si Y, Xu Y, Chen L. End-to-end single-stranded DNA sequence design with all-atom structure reconstruction. 2025. https://doi.org/10.64898/2025.12.05.692525.
The conformation of the five-membered sugar ring in DNA/RNA structures can be characterized by the five consecutive endocyclic torsion angles (see Figure below), i.e.,
ν0: C4′-O4′-C1′-C2′
ν1: O4′-C1′-C2′-C3′
ν2: C1′-C2′-C3′-C4′
ν3: C2′-C3′-C4′-O4′
ν4: C3′-C4′-O4′-C1′

Due to the ring constraint, the conformation can be characterized approximately by 5-3=2 parameters. Using the concept of pseudorotation of the sugar ring, the two parameters are the amplitude (τm) and phase angle (P).
One set of widely used formula to convert the five torsion angles to the pseudorotation parameters is due to Altona & Sundaralingam (1972): Conformational Analysis of the Sugar Ring in Nucleosides and Nucleotides. A New Description Using the Concept of Pseudorotation [J. Am. Chem. Soc., 94(23), pp. 8205–8212]. The concept is easily illustrated with an example — here with the G4 sugar ring on chain A of the Dickerson dodecamer (1bna), using Octave/Matlab code:
# xyz coordinates of the G4 sugar ring on chain A of 1bna
# ATOM 63 C4' DG A 4 21.393 16.960 18.505 1.00 53.00
# ATOM 64 O4' DG A 4 20.353 17.952 18.496 1.00 38.79
# ATOM 65 C3' DG A 4 21.264 16.229 17.176 1.00 56.72
# ATOM 67 C2' DG A 4 20.793 17.368 16.288 1.00 40.81
# ATOM 68 C1' DG A 4 19.716 17.901 17.218 1.00 30.52
# endocyclic torsion angles:
v0 = -26.7; v1 = 46.3; v2 = -47.1; v3 = 33.4; v4 = -4.4;
Pconst = sin(pi/5) + sin(pi/2.5); # 1.5388
P0 = atan2(v4 + v1 - v3 - v0, 2.0 * v2 * Pconst); # 2.9034
tm = v2 / cos(P0) # amplitude: 48.469
P = 180/pi * P0 # phase angle: 166.35 [P + 360 if P0 < 0]
The Altona & Sundaralingam (1972) pseudorotation parameters are what have been adopted in 3DNA. The Curves+ program, on the other hand, uses another set of formula due to Westhof & Sundaralingam (1983): A Method for the Analysis of Puckering Disorder in Five-Membered Rings: The Relative Mobilities of Furanose and Proline Rings and Their Effects on Polynucleotide and Polypeptide Backbone Flexibility. [J. Am. Chem. Soc., 105(4), pp. 970–976]. The two sets of formula — Altona & Sundaralingam (1972) and Westhof & Sundaralingam (1983) — give slightly different numerical values for the two pseudorotation parameters (see below).
Since Curves+ and 3DNA are currently the most commonly used programs for conformational analysis of nucleic acid structures, the subtle differences in these two pseudorotation parameters may cause confusions for users who use both programs. With the same G4 (on chain A of 1bna) sugar ring, here is the Octave/Matlab script showing how Curve+ calculates the pseudorotation parameters:
# xyz coordinates of the G4 sugar ring on chain A of 1bna
# endocyclic torsion angles, same as above
v0 = -26.7; v1 = 46.3; v2 = -47.1; v3 = 33.4; v4 = -4.4;
v = [v2, v3, v4, v0, v1]; # reorder them into vector v[]
A = 0; B = 0;
for i = 1:5
t = 0.8 * pi * (i - 1);
A += v(i) * cos(t);
B += v(i) * sin(t);
end
A *= 0.4; # -48.476
B *= -0.4; # 11.516
tm = sqrt(A * A + B * B); # 49.825
c = A/tm; s = B/tm;
P = atan2(s, c) * 180 / pi; # 166.64
For this specific example, i.e., the G4 sugar ring on chain A of 1bna, the pseudorotation parameters as calculated by 3DNA following Altona & Sundaralingam (1972) and Curves+ following Westhof & Sundaralingam (1983) are as follows:
|
amplitude (τm) |
phase angle (P) |
| 3DNA |
48.469 |
166.35 |
| Curves+ |
49.825 |
166.64 |
Needless to say, for the majority of cases like the one shown here, the differences are subtle; very few people would notice them or be bothered at all. For those who do care about such little details, however, this post shows where the discrepancies really come from.

In the field of nucleic acid structural analysis, it seems fair to say that Curves+ and 3DNA are nowadays the top two choices. To the best of my knowledge, these two programs are also the only ones that confirm to the standard base reference frame. Moreover, as noted in my previous post, Curves+ and 3DNA are “constructive competitors” with complementary functionality: Curves+ is unique in providing a curvilinear helical axis, a bending analysis, a full description of groove widths and depths and its seamless integration to the analysis of molecular dynamics trajectories, while 3DNA’s strength lies in its cohesrent approach combining analysis, rebuilding, and visualization into one package.
Given the complementarity between Curve+ and 3DNA, it makes sense to build a ‘bridge’ between the two so users can easily take advantage of both programs. Starting from 3DNA v1.5, find_pair has the -c option to generate input for Curves directly from a PDB file. Over the years, this option appears to have received little attention — at least, I am not aware of any literature reference to it. Now, the updated Curves+ program has introduced the new lib name list variable, among other changes. I have thus added the -curves+ option (abbreviation -c+) to find_pair to make its output compatible with Curves+.
As always, the point/process is best illustrated with an example — here with the Dickerson B-DNA dodecamer solved at high resolution by Williams et al. (PDB entry 355d).
find_pair -c+ 355d.pdb 355d-curves+.inp
The generated file 355d-curves+.inp has the following content:
&inp file=355d.pdb,
lis=355d,
fit=.t.,
lib=/Users/xiangjun/Curves+/standard,
isym=1,
&end
2 1 -1 0 0
1 2 3 4 5 6 7 8 9 10 11 12
24 23 22 21 20 19 18 17 16 15 14 13
which can be fed into Curves+ as below,
Cur+ < 355d-curves+.inp
The four output files are: 355d.cda, 355d.lis, 355d_X.pdb and 355d_b.pdb.
Please note the followings:
- The environment variable CURVES_PLUS_STDLIB should be set, pointing to the directory where Curves+ is installed (containing files
standard_b.lib and standard_s.lib). In the example above, CURVES_PLUS_STDLIB is set to /Users/xiangjun/Curves+.
- The
find_pair -c+ option (currently) is applicable only to double helical DNA/RNA structures, the most common application scenario.
- The
-c+ option ignores HETATM records, in accordance with Curves+ where proteins, water and HETATM are automatically removed at input. (see Curves+ user manual, section Input data)
- To run
Cur+ < 355d-curves+.inp again in the same folder, the four output files must first be deleted (e.g., rm -f 355d.cda 355d.lis 355d_[Xb].pdb). This is best taken care of via a script.
Obviously, the nucleic acid structure community benefits the most to have both Curves+ and 3DNA at its disposal and be able to easily switch between them — hopefully, the find_pair -c+ option would serve as such a ‘bridge’.
From the very beginning, 3DNA calculates a set of nucleic acid backbone parameters, including the six main chain torsion angles (α, β, γ, δ, ε, and ζ) around the covalent bonds, χ about the glycosidic bond, and the sugar pucker (see figure below). For double helical structures, the standard analyze output (.out file) has a section for “Main chain and chi torsion angles,” and another dedicated to “Sugar conformational parameters”. Based on my experience/understanding, these two parts are well recognized and utlizied by 3DNA users. What has receive little attention (in spite of the several posts I’ve written on the topic), though, is 3DNA’s applicability to single-stranded (ss) RNA structures for the backbone torsions, among other parameters. Using the fully refined crystal structure of the Haloarcula marismortui large ribosomal subunit (PDB entry 1jj2) as an example, the procedure is below:
find_pair -s 1jj2.pdb 1jj2.nts
analyze 1jj2.nts
# or the above two steps can be combined:
find_pair -s 1jj2.pdb stdout | analyze stdin
# see output file '1jj2.outs'
In retrospect, the fact that 3DNA has been little used for RNA backbone conformational analysis is of no surprise:
- While base-pair parameters have different (oftentimes confusing) definitions, these backbone parameters are pretty “standard” — thus, for example, any program for DNA/RNA structural analysis would give the same numerical values for α or χ torsion angles.
- The two-step process as illustrated above is a bit awkward, and the torsions are “buried” among many other parameters.
- 3DNA is more directly “linked” (conceivably) to DNA base pairs than to RNA backbone.
So while adapting the Zp parameter for ss DNA/RNA structures in 3DNA v2.1, I also take this opportunity to add the -torsion option to analyze with the following handy features:
- Streamline the calculation by starting directly from a PDB file and output only backbone parameters. So the above example can be shortened to
analyze -t=1jj2.tor 1jj2.pdb; the output file is named 1jj2.tor.
- Classify backbone into BI/BII conformation, and base χ into syn / anti.
- Add pseudo-torsions, and Zp and Dp as defined by Richardson et al.
- Handle pseudouridine sensibly, and work also for nucleic acid structure with only backbone atoms.
- Be easy to use, efficient and robust — it takes ~1 second to process the large ribosomal subunit 1jj2 (with 2876 nucleotides consisting of 23S rRNA and 5S rRNA) on my MacBook Air.
Overall, analyze -torsion is designed to be pragmatic and allows for automatic processing of all NDB entries or molecular dynamics trajectories. Given below is an excerpt of the three sections from an analyze -torsion run on 1jj2:
****************************************************************************
Main chain and chi torsion angles:
Note: alpha: O3'(i-1)-P-O5'-C5'
beta: P-O5'-C5'-C4'
gamma: O5'-C5'-C4'-C3'
delta: C5'-C4'-C3'-O3'
epsilon: C4'-C3'-O3'-P(i+1)
zeta: C3'-O3'-P(i+1)-O5'(i+1)
chi for pyrimidines(Y): O4'-C1'-N1-C2
chi for purines(R): O4'-C1'-N9-C4
If chi is in range [-90, +90], syn conformation
otherwise, it is in anti conformation
e-z: epsilon - zeta
BI: e-z = [-160, +20]
BII: e-z = [+20, +200]
base chi alpha beta gamma delta epsilon zeta e-z
1 0:..10_:[..U]U -62.5(syn) --- --- 56.2 74.0 142.2 -87.8 -130.1(BI)
2 0:..11_:[..A]A 171.5(anti) 173.2 -161.0 168.5 84.0 -112.1 -65.4 -46.7(BI)
3 0:..12_:[..U]U -167.7(anti) -70.7 168.4 53.0 78.5 -128.5 -46.4 -82.1(BI)
4 0:..13_:[..G]G -172.5(anti) -61.8 170.4 67.7 73.5 -166.7 -79.6 -87.1(BI)
5 0:..14_:[..C]C -166.0(anti) -73.0 -172.5 55.1 83.2 -143.3 -77.7 -65.6(BI)
6 0:..15_:[..C]C -155.5(anti) -60.9 174.1 47.3 80.3 -154.4 -71.2 -83.2(BI)
****************************************************************************
Pseudo (virtual) eta/theta torsion angles:
Note: eta: C4'(i-1)-P(i)-C4'(i)-P(i+1)
theta: P(i)-C4'(i)-P(i+1)-C4'(i+1)
eta': C1'(i-1)-P(i)-C1'(i)-P(i+1)
theta': P(i)-C1'(i)-P(i+1)-C1'(i+1)
eta": Borg(i-1)-P(i)-Borg(i)-P(i+1)
theta": P(i)-Borg(i)-P(i+1)-Borg(i+1)
base eta theta eta' theta' eta" theta"
1 0:..10_:[..U]U --- --- --- --- --- ---
2 0:..11_:[..A]A -174.6 -129.7 177.0 -127.7 -157.5 -75.5
3 0:..12_:[..U]U 149.1 -105.1 174.1 -101.2 -111.0 -69.4
4 0:..13_:[..G]G 169.0 -172.5 -156.6 -169.2 -93.3 -137.1
5 0:..14_:[..C]C 176.2 -143.4 179.6 -140.6 -144.6 -120.6
6 0:..15_:[..C]C 165.0 -147.7 177.4 -146.8 -149.2 -121.7
****************************************************************************
Sugar conformational parameters:
Note: v0: C4'-O4'-C1'-C2'
v1: O4'-C1'-C2'-C3'
v2: C1'-C2'-C3'-C4'
v3: C2'-C3'-C4'-O4'
v4: C3'-C4'-O4'-C1'
tm: the amplitude of pucker
P: the phase angle of pseudorotation
Zp: z-coordinate of the 3' phosphorus atom (P) expressed in the
standard base reference frame; it's POSITIVE when P is on
the +z-axis side (base in anti conformation); NEGATIVE if
P is on the -z-axis side (base in syn conformation)
Dp: perpendicular distance of the 3' P atom to the glycosydic bond
[as per the MolProbity paper of Richardson et al. (2010)]
base v0 v1 v2 v3 v4 tm P Puckering Zp Dp
1 0:..10_:[..U]U -11.3 -15.4 34.5 -41.8 33.5 41.6 33.8 C3'-endo -0.13 3.53
2 0:..11_:[..A]A 11.4 -30.2 36.9 -31.5 12.6 36.9 1.2 C3'-endo 4.74 4.78
3 0:..12_:[..U]U 3.6 -29.3 42.4 -41.3 23.8 43.6 13.9 C3'-endo 4.67 4.82
4 0:..13_:[..G]G -13.0 -17.8 39.8 -47.9 38.5 47.8 33.7 C3'-endo 4.45 4.46
5 0:..14_:[..C]C 6.0 -28.4 38.9 -36.5 19.2 39.5 10.1 C3'-endo 4.57 4.70
6 0:..15_:[..C]C 1.9 -26.4 39.6 -39.6 23.7 41.2 16.0 C3'-endo 4.32 4.61
Given the x-, y-, and z-coordinates of four points (a-b-c-d) in 3-dimensional (3D) space, how to calculate the torsion angle? Overall, this is a well-solved problem in structural biology and chemistry; one can find a description of torsion angle in many text books and on-line documents. The algorithm for its calculation is implementated in virtually every software package in computational structural biology and chemistry.
As basic as the concept is, however, it is important (based on my experience) to have a clear understanding of how torsion angle is defined in order to really get into the 3D world. Here is a worked example using Octave/Matlab of my simplified, geometry-based implementation of calculating torsion angle, including how to determine its sign. No theory or (complicated) mathematical formula, just a step-by-step illustration of how I solve this problem.
- Coordinates of four points are given in variable
abcd:
abcd = [ 21.350 31.325 22.681
22.409 31.286 21.483
22.840 29.751 21.498
23.543 29.175 22.594 ];
- Two auxiliary functions: norm_vec() to normalize a vector; get_orth_norm_vec() to get the orthogonal component (normalized) of a vector with reference to another vector, which should have already been normalized.
function ovec = norm_vec(vec)
ovec = vec / norm(vec);
endfunction
function ovec = get_orth_norm_vec(vec, vref)
temp = vec - vref * dot(vec, vref);
ovec = norm_vec(temp);
endfunction
- Get three vectors: b_c is the normalized vector b→c; b_a_orth is the orthogonal component (normalized) of vector b→a with reference to b→c; c_d_orth is similarly defined, as the orthogonal component (normalized) of vector c→d with reference to b→c.
b_c = norm_vec(abcd(3, :) - abcd(2, :))
% [0.2703158 -0.9627257 0.0094077]
b_a_orth = get_orth_norm_vec(abcd(1, :) - abcd(2, :), b_c)
% [-0.62126 -0.16696 0.76561]
c_d_orth = get_orth_norm_vec(abcd(4, :) - abcd(3, :), b_c)
% [0.41330 0.12486 0.90199]
- Now the torsion angle is defined as the angle between the two vectors, b_a_orth and c_d_orth, and can be easily calculated by their dot product. The sign of the torsion angle is determined by the relative orientation of the cross product of the same two vectors with reference to the middle vector b→c. Here they are in opposite direction, thus the torsion angle is negative.
angle_deg = acos(dot(b_a_orth, c_d_orth)) * 180 / pi % 65.609
sign = dot(cross(b_a_orth, c_d_orth), b_c) % -0.91075
if (sign < 0)
ang_deg = -angle_deg % -65.609
endif
A related concept is the so-called dihedral angle, or more generally the angle between two planes. As long as the normal vectors to the two corresponding planes are defined, the angle between them is easy to work out.
It’s worth noting that the helical twist angle in SCHNAaP and 3DNA is calculated similarly.
Backbone conformation of nucleic acid structures is most characterized by a set of 6 torsion angles (α, β, γ, δ, ε, and ζ) around the consecutive chemical bonds, chi (χ) quantifying the relative base/sugar orientation, plus the sugar pucker.

This large number of DNA/RNA backbone conformational parameters is in striking contrast to the two torsion angles (φ and ψ) in protein structures, routinely employed in Ramachandran plot. Over the years, the nucleic acid community has come up with simplified ways to represent DNA/RNA backbone conformation. Thus far, the most widely used one is the pseudo-torsion angles (See figure below) η: C4′(i-1)-P(i)-C4′(i)-P(i+1) and θ: P(i)-C4′(i)-P(i+1)-C4′(i+1).

The history of the P—C4′ virtual-bond concept and its application in RNA structure analysis have recently been reviewed by Pyle et al. in A new way to see RNA [Q Rev Biophys. 2011, 44(4), 433—466], where the following three contributions are highlighted:
- Olson (1980). Configurational statistics of polynucleotide chains. An updated virtual bond model to treat effects of base stacking., Macromolecules 13(3), 721—728.
- Malathi & Yathindra (1980). A novel virtual bond scheme to probe ordered and random coil conformations of nucleic acids: Configurational statistics of polynucleotide chains. Current Science, 49, 803—807.
- Duarte & Pyle (1998). Stepping through an RNA structure: A novel approach to conformational analysis. Journal of Molecular Biology, 284, 1465—1478.
More recently, Pyle et al. also employed a modified version of the pseudo-torsions, η′: C1′(i-1)-P(i)-C1′(i)-P(i+1) and θ′: P(i)-C1′(i)-P(i+1)-C1′(i+1), i.e., using C1′ instead of C4′, and found that:
The η′ and θ′ torsions are more suitable when interpreting crystallographic density because the C1′ atom is covalently bound to the nucleoside base and therefore can be more easily and accurately located within a low-resolution map.
While implementing the -torsion option to analyze to make it more explicit that 3DNA readily calculates conventional backbone torsion angles, I also take this opportunity to add the pseudo-torsion angles — η/θ and η′/θ′, among other new parameters. Moreover, while I am at it, I cannot help but also compute yet another set of pseudo-torsion angles: η″/θ″. Here, instead of C1′ or C4′, the origin of the base reference frame is employed; it can be taken as a _pseudo_-atom more accurately defined by the base plane than any real single atom.
The usefulness of η″/θ″, especially in comparison with η/θ and η′/θ′, remains to be determined. However, only η″/θ″ uniquely takes advantage of the two most accurately determined entities in a nucleic acid structure, the heavy phosphorus atom and the rigid base plane [see discussion (p.16) in the Richardson et al. MolProbity paper, Acta Cryst. (2010). D66, 12–21] Presumably, η″/θ″ provides a new perspective in RNA structural analysis by combining the backbone and the base.
Here is the pseudo-torsions for the yeast phenylalanine transfer RNA (6tna by simply running analyze -torsion=6tna.tor 6tna.pdb):
Pseudo (virtual) eta/theta torsion angles:
Note: eta: C4'(i-1)-P(i)-C4'(i)-P(i+1)
theta: P(i)-C4'(i)-P(i+1)-C4'(i+1)
eta': C1'(i-1)-P(i)-C1'(i)-P(i+1)
theta': P(i)-C1'(i)-P(i+1)-C1'(i+1)
eta": Borg(i-1)-P(i)-Borg(i)-P(i+1)
theta": P(i)-Borg(i)-P(i+1)-Borg(i+1)
base eta theta eta' theta' eta" theta"
1 A:...1_:[..G]G --- -126.6 --- -141.5 --- -130.4
2 A:...2_:[..C]C 167.8 -168.3 174.6 -152.5 -151.4 -115.4
3 A:...3_:[..G]G 160.4 -119.8 -171.9 -138.9 -123.6 -119.2
4 A:...4_:[..G]G 148.0 -164.2 162.1 -159.2 -154.4 -124.6
5 A:...5_:[..A]A 168.7 -137.6 -175.9 -137.8 -129.5 -115.0
6 A:...6_:[..U]U 171.8 -145.7 -172.5 -140.5 -131.3 -124.7
7 A:...7_:[..U]U -151.0 -47.8 -136.0 -58.6 -117.7 -30.2
8 A:...8_:[..U]U 160.9 159.7 -161.0 -163.6 -144.2 178.0
9 A:...9_:[..A]A -137.0 -48.6 -158.1 -108.9 161.5 -104.7
10 A:..10_:[2MG]g 33.1 -135.8 93.4 -134.6 134.1 -113.0
11 A:..11_:[..C]C 167.2 -138.3 -179.4 -137.7 -142.4 -118.7
12 A:..12_:[..U]U 165.5 -120.7 -179.3 -128.0 -145.8 -106.7
13 A:..13_:[..C]C 174.1 -173.6 -165.5 179.6 -120.9 -180.0
14 A:..14_:[..A]A 173.0 -144.0 172.7 -132.4 177.6 -72.7
15 A:..15_:[..G]G 154.7 110.6 -176.2 85.5 -97.7 -76.9
16 A:..16_:[H2U]u 76.3 94.1 65.3 119.7 -152.8 -123.8
17 A:..17_:[H2U]u -36.7 -79.6 -50.7 -136.6 -142.7 -159.0
18 A:..18_:[..G]G -9.7 -166.8 41.7 -158.6 28.9 -120.4
19 A:..19_:[..G]G -131.6 -35.8 -122.9 -67.8 -104.3 -10.5
20 A:..20_:[..G]G 160.9 -93.2 -161.6 -98.9 -174.1 -112.3
21 A:..21_:[..A]A -83.6 152.5 -72.8 155.7 -59.1 155.4
22 A:..22_:[..G]G 164.1 169.4 160.0 -178.5 159.1 -157.6
23 A:..23_:[..A]A 177.6 -148.5 -174.5 -142.7 -154.5 -114.3
24 A:..24_:[..G]G 167.2 -98.9 -171.7 -128.6 -127.6 -99.1
25 A:..25_:[..C]C 151.6 -153.5 167.3 -140.8 -137.7 -84.8
26 A:..26_:[M2G]g 156.2 -137.4 -175.2 -135.2 -100.0 -104.2
27 A:..27_:[..C]C 166.2 -145.5 -177.9 -140.4 -129.1 -116.8
28 A:..28_:[..C]C 164.7 -140.5 175.8 -145.3 -152.7 -123.4
29 A:..29_:[..A]A 161.2 -145.3 175.7 -144.9 -142.0 -126.0
30 A:..30_:[..G]G -173.5 -120.3 -158.4 -133.2 -126.6 -94.4
31 A:..31_:[..A]A 169.8 -153.1 177.7 -140.4 -124.5 -81.5
32 A:..32_:[OMC]c 154.4 -126.8 -178.7 -131.3 -104.1 -128.0
33 A:..33_:[..U]U 170.0 -103.9 -179.9 -152.7 -164.6 143.6
34 A:..34_:[OMG]g -4.7 -123.7 41.8 -124.8 31.6 -99.6
35 A:..35_:[..A]A 163.5 -104.3 176.9 -127.9 -137.5 -128.2
36 A:..36_:[..A]A 175.9 173.6 180.0 -167.7 -156.4 -118.3
37 A:..37_:[.YG]g 166.8 -131.7 -174.5 -133.0 -115.1 -82.9
38 A:..38_:[..A]A 167.7 -121.6 -175.7 -114.3 -109.9 -79.9
39 A:..39_:[PSU]P 168.3 -146.8 -160.2 -146.4 -98.6 -116.5
40 A:..40_:[5MC]c 160.6 -138.7 174.0 -141.8 -139.7 -126.5
41 A:..41_:[..U]U 164.8 -161.4 175.9 -152.3 -150.5 -117.6
42 A:..42_:[..G]G 174.3 -140.9 -170.3 -145.4 -129.1 -121.3
43 A:..43_:[..G]G 169.6 -159.0 -176.2 -154.9 -133.7 -133.1
44 A:..44_:[..A]A 174.0 -121.5 -174.2 -122.0 -143.1 -74.9
45 A:..45_:[..G]G 174.4 -132.5 -166.2 -128.1 -101.8 -128.9
46 A:..46_:[7MG]g -112.8 -113.4 -127.2 -138.3 -139.8 -152.1
47 A:..47_:[..U]U -63.2 -53.8 -1.1 -92.0 22.8 -124.7
48 A:..48_:[..C]C -84.7 59.6 -20.1 8.9 19.3 -104.5
49 A:..49_:[5MC]c -56.8 -140.1 -29.9 -143.6 98.1 -125.4
50 A:..50_:[..U]U 173.6 -146.4 -178.3 -140.6 -147.6 -117.8
51 A:..51_:[..G]G 160.8 -148.1 -178.6 -150.7 -140.7 -121.9
52 A:..52_:[..U]U 164.9 -144.0 175.8 -143.5 -139.9 -114.3
53 A:..53_:[..G]G 168.2 -140.9 -171.1 -144.0 -121.6 -117.3
54 A:..54_:[5MU]u 167.0 -131.1 178.3 -124.9 -139.9 -77.0
55 A:..55_:[PSU]P 167.6 -114.2 -172.8 -155.6 -113.0 146.0
56 A:..56_:[..C]C 35.0 -121.5 52.6 -126.2 26.5 -83.8
57 A:..57_:[..G]G 168.4 -148.1 -177.1 -131.1 -115.4 -111.7
58 A:..58_:[1MA]a -136.3 -133.3 -106.5 -176.7 -105.3 149.6
59 A:..59_:[..U]U 23.0 -130.9 33.0 -115.4 48.2 -68.2
60 A:..60_:[..C]C -163.6 -54.3 -123.2 -76.4 -79.6 -36.4
61 A:..61_:[..C]C 125.5 -153.3 169.7 -144.7 -153.8 -123.4
62 A:..62_:[..A]A 172.5 -139.3 -177.0 -137.6 -150.7 -114.6
63 A:..63_:[..C]C 165.8 -146.6 -178.5 -149.8 -139.2 -127.8
64 A:..64_:[..A]A 164.7 -144.9 176.5 -145.8 -145.3 -118.1
65 A:..65_:[..G]G 170.4 -152.3 -175.5 -151.5 -132.3 -122.1
66 A:..66_:[..A]A 168.0 -152.0 -177.4 -150.2 -133.0 -118.7
67 A:..67_:[..A]A 170.9 -141.8 -178.4 -140.4 -134.8 -123.1
68 A:..68_:[..U]U 164.8 -135.1 -178.9 -137.9 -143.7 -95.2
69 A:..69_:[..U]U 168.2 -154.9 -174.3 -157.1 -112.2 -144.8
70 A:..70_:[..C]C 160.6 -153.2 170.7 -153.5 -164.4 -125.1
71 A:..71_:[..G]G 161.8 -144.3 172.1 -143.1 -145.7 -124.2
72 A:..72_:[..C]C 176.7 -136.4 -169.3 -134.5 -134.9 -87.1
73 A:..73_:[..A]A 160.6 -142.8 -179.7 -139.7 -112.8 -104.4
74 A:..74_:[..C]C -176.9 -115.9 -163.1 -115.4 -117.2 -68.7
75 A:..75_:[..C]C 169.8 80.9 -170.0 74.9 -108.5 -91.3
76 A:..76_:[..A]A --- --- --- --- --- ---
In nucleic acid structures, the chi (χ) torsion angle is about the glycosidic bond (N-C1′) that connects the sugar and the A/C/G/T/U bases (or their modified variants). Specifically, for pyrimidines (C, T and U), χ is defined by O4′-C1′-N1-C2; and for purines (A and G) by O4′-C1′-N9-C4 (see figure below).

Pseudouridine (5-ribosyluracil, PSU) was the first identified modified nucleoside in RNA and is the most abundant. PSU is unique in that it has a C-glycosidic bond (C-C1′) instead of the N-glycosidic bond common to all other nucleosides, canonical or modified. It thus poses a problem as to how to calculate the χ torsion angle: should it be O4′-C1′-C5-C4, reflecting the actual glycosidic bond connection, or should the conventional definition O4′-C1′-N1-C2 still be applied literally? As a concrete example, the figure below shows the (slightly) different numerical values (–162.7° vs. –163.9°), as given by the two definitions, for PSU 6 on chain A of the PDB entry 3cgp (based on the 2009 Biochemistry article by Lin & Kielkopf titled X-ray structures of U2 snRNA-branchpoint duplexes containing conserved pseudouridines).
Needless to say, the specific definition of the χ torsion angle for PSU in RNA structures is a very subtle point, and I am not aware of any discussion on this issue in literature. In 3DNA, PSU is identified explicitly, and χ is defined by O4′-C1′-C5-C4. In NDB and a couple of other tools I am familiar with, χ for PSU is defined by O4′-C1′-N1-C2. Again using 3cgp (figure below) as an example, 3DNA gives –162.7°, whilst NDB gives –163.9°. Additionally, this distinction in N-C1′ vs. C-C1′ connection also comes into play when calculating the perpendicular distance from the 3′ phosphorus atom to the glycosidic bond, as per Richardson et al.
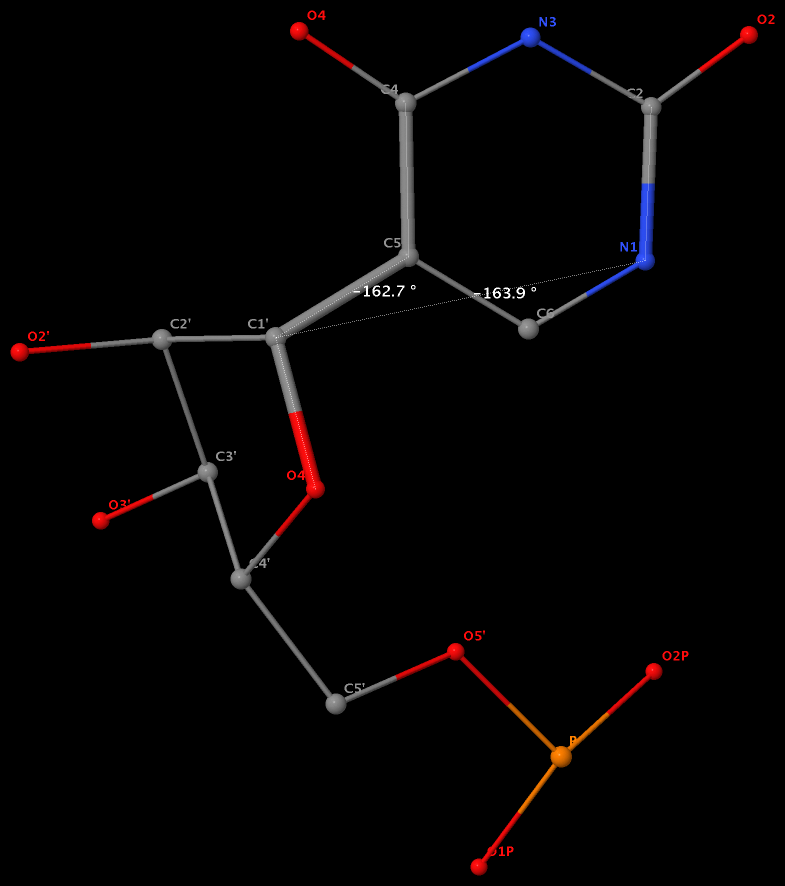
Except for pseudouridine, a nucleoside in DNA/RNA contains an N-glycosidic bond that connects the base to the sugar. The chi (χ) torsion angle, which characterizes the relative base/sugar orientation, is defined by O4′-C1′-N1-C2 for pyrimidines (C, T and U), and O4′-C1′-N9-C4 for purines (A and G).
Normally (as in A- and B-form DNA/RNA duplex), χ falls into the ranges of +90° to +180°; –90° to –180° (or 180° to 270°), corresponding to the anti conformation (Figure below, top). Occasionally, χ has values in the range of –90° to +90°, referring to the syn conformation (Figure below, bottom). Note that in left-handed Z-DNA with CG repeating sequence, the purine G is in syn conformation whilst the pyrimidine C is anti.
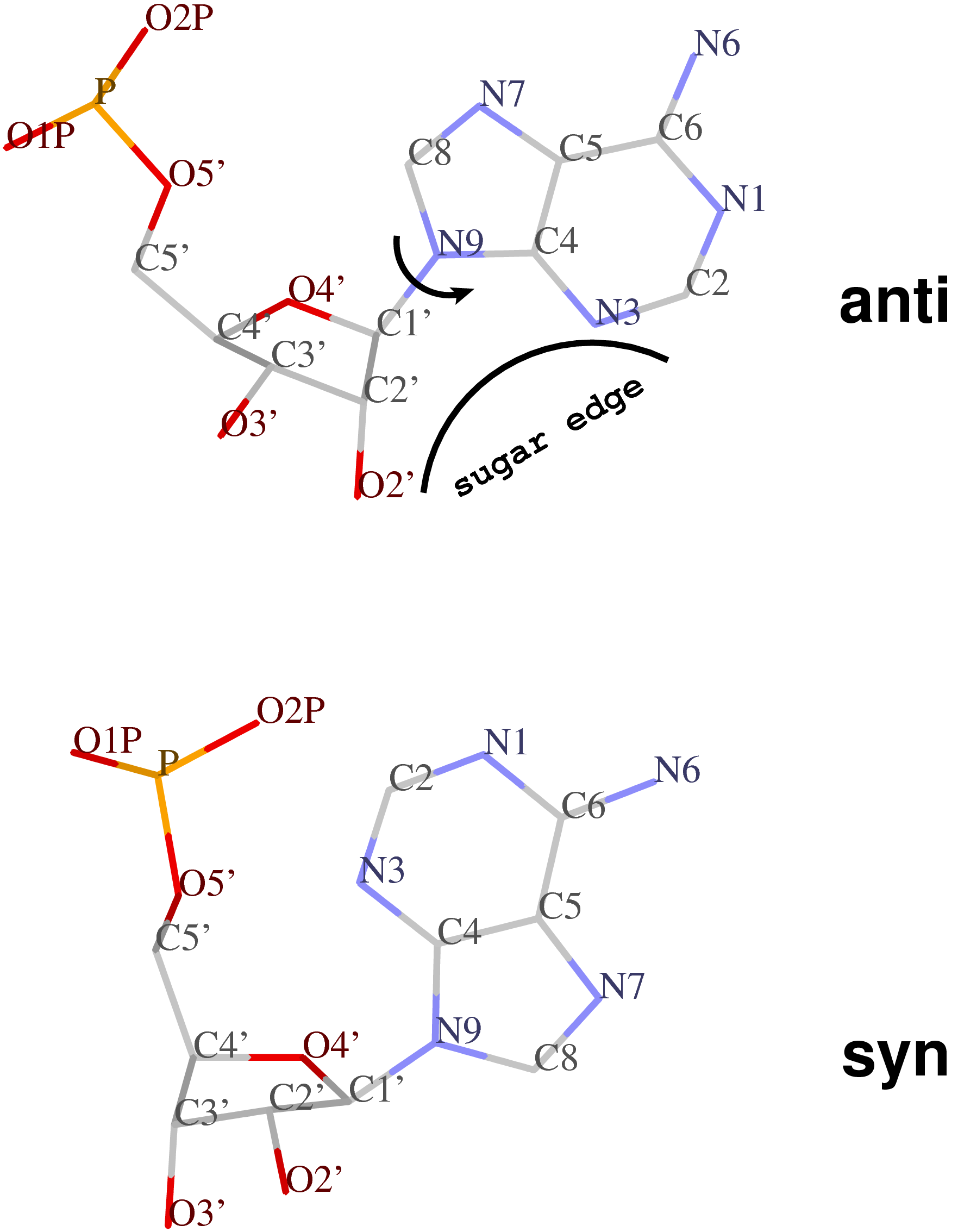
Presumably, the χ-related anti / syn conformation is a simple geometric concept. Nevertheless, the N-glycosidic bond and the corresponding χ torsion angle illustrate that the base and the sugar are two separate entities, i.e. there is an internal degree of freedom between them. In this respect, it is worth noting that the Leontis-Westhod sugar edge for base-pair classification corresponds to the anti form (as applied to RNA) only. When a base is flipped over into the syn conformation, the “sugar edge”, defined in connection with the minor (shallow) groove side of a nitrogenous bases, simply does not exist.
Base-flipping (anti / syn conformation switch) is one of the factors associated with the two possible relative orientations of the two bases in a pair, characterized explicitly in 3DNA as of type M+N or M–N since the 2003 NAR paper (Figure 2, linked below). I re-emphasized this distinction in our 2010 GpU dinucleotide platform paper (in particular, see supplementary Figure S2). Unfortunately, this subtle (but crucial, in my opinion) point has never been taken seriously (or at all) by the RNA community, even with 3DNA’s wide adoption. However, as people know 3DNA deeper/better and take RNA base-pair classification more rigorously, I have no doubt that the simplicity of this explicit distinction and the resultant full quantification of each and every possible base pair using standard geometric parameters will gradually be appreciated.

As of 3DNA v2.1, the output of the χ torsion angle is also associated with its classification in anti / syn conformation, among other new features (see for example the output for 6tna).
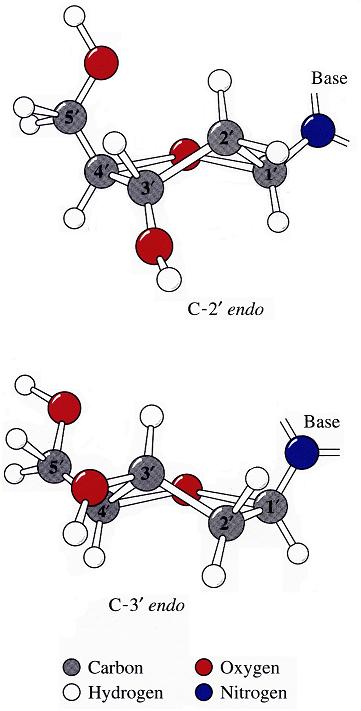
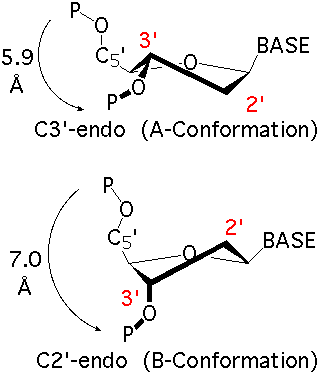
The sugar puckers in DNA/RNA structures are predominately in either C3′-endo (A-DNA or RNA) or C2′-endo (B-DNA; see Figure below, left), corresponding to the A- or B-form conformation in a duplex. In these two sugar conformations, the distance between neighboring phosphorus (P) atoms and the orientation of P relative to the sugar/bases are also dramatically different (figure below, right).
Recently, I carefully re-read some articles on RNA backbone conformation by Richardson et al., including:
I became intrigued by one of their observations: i.e., the correlation between the sugar pucker and a simple distance parameter:
C3′-endo and C2′-endo sugar puckers are highly correlated to the perpendicular distance between the C1′–N1/9 glycosidic bond vector and the following phosphate: > 2.9 Å for C3′-endo and < 2.9 Å for C2′-endo. (p.16 from the MolProbity paper).
Out of curiosity and for a better understanding of this correlation, I played around with some sample cases both visually and numerically. Overall, this involves a simple geometric calculation, i.e., the shortest distance from a point to a line in three-dimensional space. Given below is the Octave/Matlab script for calculating the distances for G175 and U176 of PDB entry 1jj2 (the large ribosomal subunit of Haloarcula marismortui):
function d = get_p3_nc_dist(P3, C1, N)
C1_N = N - C1; # vector from C1′ to N
nv_C1_N = C1_N / norm(C1_N); # normalized vector
C1_P3 = P3 - C1; # vector from C1′ to P3
proj = dot(C1_P3, nv_C1_N);
d = norm(C1_P3 - proj * nv_C1_N);
end
## G175
P3 = [70.104 112.366 44.586];
C1 = [73.017 109.666 45.304];
N9 = [74.445 109.380 45.288];
d1 = get_p3_nc_dist(P3, C1, N9) # 2.2 Å -- C2′-endo
## U176
P3 = [66.871 116.402 46.804];
C1 = [68.213 112.454 49.279];
N1 = [69.678 112.480 49.438];
d2 = get_p3_nc_dist(P3, C1, N1) # 4.6 Å -- C3′-endo
The GpU dinucleotide used in the above example forms a platform (see figure below), where the sugar of G175 adopts a C2′-endo conformation, and that of U176 C3′-endo. Indeed, the distance for G175 is 2.2 Å (< 2.9 Å); whilst the value for U176 is 4.6 Å (> 2.9 Å).

Note that the Richardson et al. articles focus on the RNA backbone, without paying attention to the base (pair) geometry. The 3DNA Zp parameter, which is the mean z-coordinate of the two P atoms in the mean reference frame of a dinucleotide step (see figure below), has been readily adapted to single-stranded RNA structures. For example, the vertical distances of the 3′ P atoms to the G175 and U176 base planes are 1.9 Å and 4.4 Å, respectively. Since base planes and the P atoms are the two most accurately located entities in a given nucleic acid structure, the nucleotide-based Zp variant is presumably more robust and discriminative than the distance from P to the glycosidic bond.

This new single-stranded based “Zp” parameter is available as of 3DNA v2.1.
RNA has three salient structural features (compared to DNA): it contains the ribose (not deoxyribose) sugar, it has the uracil (not thymine) base, and it is normally single (not double)-stranded. The O2′(G)…O2P(U) H-bond stabilized GpU dinucleotide platform may turn out to be the smallest unit with all those RNA hallmarks.
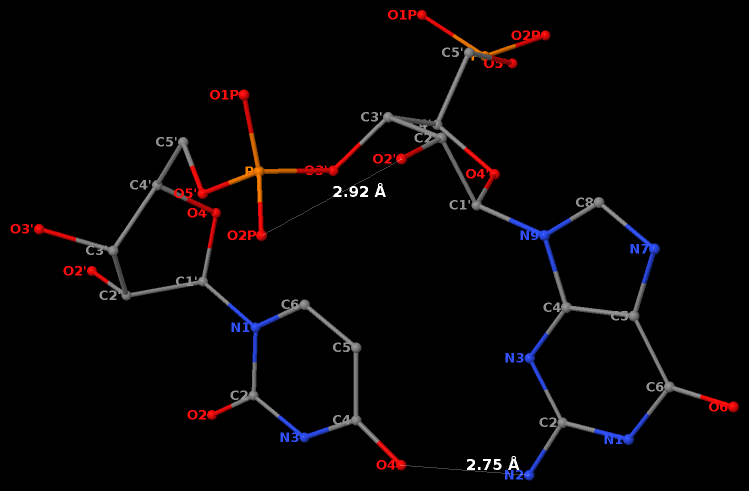
First, it must have the guanosine ribose to have the 2′-hydroxyl group form the O2′(G)…O2P(U) H-bond.
Second, the methyl group in position 5 of thymine would cause steric clash with guanosine, thus disrupting the N2(G)…O4(U) base-base H-bond to form the GpU dinucleotide platform.
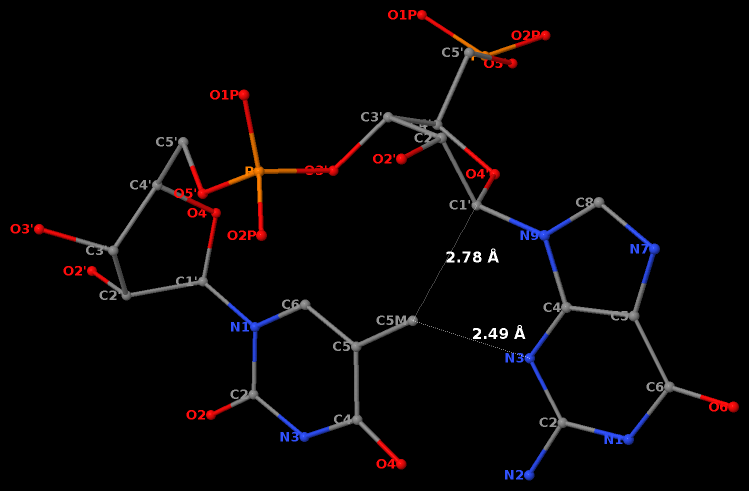
Third, a dinucleotide, by definition, is single-standed. The two H-bonds, plus the covalent linkage, makes the GpU platform extremely rigid (see Figure 1 of our 2010 NAR paper).
Moreover, the GpU platform is directional: swapping the two bases while keeping the sugar-phosphate backbone fixed does not allow for a base-base H-bond, thus no UpG dinucleotide platform.
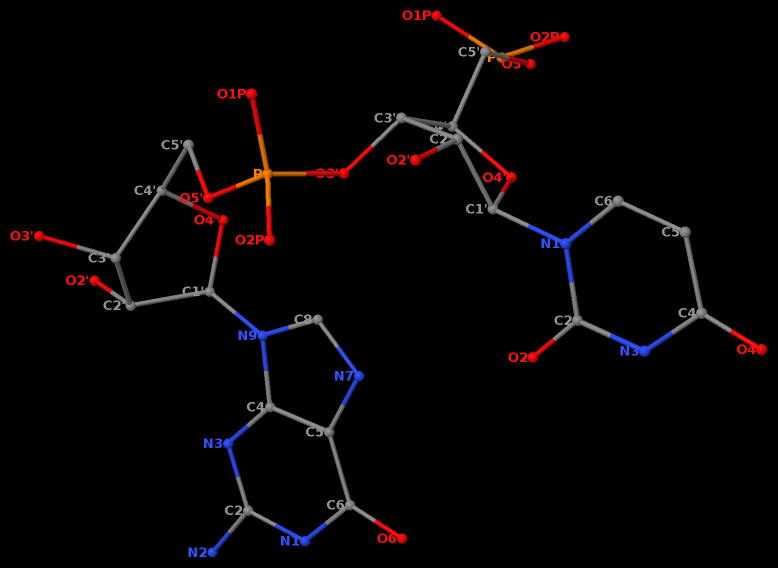
It worth noting that state-of-the-art quantum chemistry calculations have verified the importance of the O2′(G)…O2P(U) H-bond in stabilizing the GpU dinucleotide platform.